
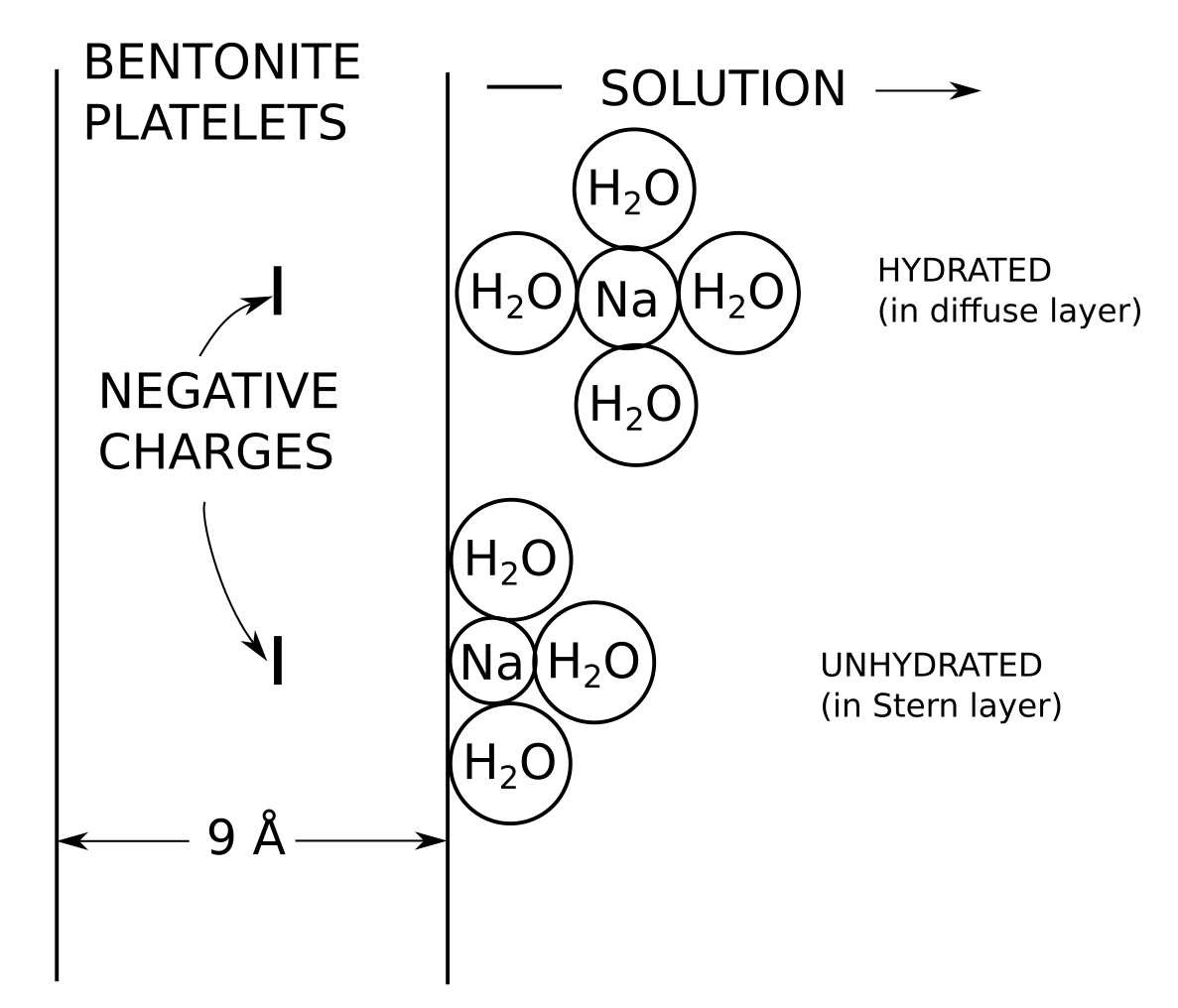
I argue that the only significant pore type in water saturated compacted bentonite is interlayers, by which I mean pores where the exchangeable cations reside (together with any other dissolved species). From this perspective it naturally follows that a homogeneous view is a suitable starting point for modeling compacted bentonite. I have presented, used, and discussed the homogeneous mixture model in many places on the blog, the main sources being
- Sorption part II: Letting go of the bulk water
- Donnan equilibrium and the homogeneous mixture model
- Sorption part IV: What is Kd?
For reasons I can’t get my head around, a homogeneous view of compacted bentonite is not the mainstream in contemporary bentonite research. Instead we are stuck with “the mainstream view”, which postulates several distinctly different pore structures within the bentonite; in particular, the mainstream view uses a bulk water phase as a starting point and also distinguishes between “outer” and “inner” basal surfaces. Electric double layers are assumed to only exist on “outer” surfaces, while the function of the “inner” basal surfaces is mostly shrouded in mystery.
On the blog I have also presented plenty of experimental support for a homogeneous view. A main argument is that the conditions for swelling pressure — the most profound feature of bentonite in equilibrium with an external solution — are essentially fulfilled automatically in the homogeneous mixture model. The mainstream view, in contrast, requires handling of the seemingly contradictory situation of having swelling pressure while the water chemical potential is supposedly restored without pressurization. Proponents of the mainstream view often deal with this by simply ignoring swelling phenomena altogether.
I have also on the blog dissected several studies that argue for a non-homogeneous view, but that actually provide evidence for the opposite when examined more carefully. Consider in particular:
- The danger of log-log plots — measuring and modeling “apparent” diffusivity
- The failure of Archie’s law validates the homogeneous mixture model
- Molecular dynamics simulations do not support complete anion exclusion
Glaus et al. (2007)
To discuss further evidence for homogeneity, we turn to one of the most profound bentonite studies published on this side of 2000: “Diffusion of \(^{22}\mathrm{Na}\) and \(^{85}\mathrm{Sr}\) in Montmorillonite: Evidence of Interlayer Diffusion Being the Dominant Pathway at High Compaction” (Glaus et al. 2007).
By systematically varying background concentration, material, and diffusing tracer, Glaus et al. (2007) clearly demonstrate, not only that the exchangeable cations are mobile, but that they dominate the flux in through-diffusion tests in highly compacted montmorillonite. While this certainly is an argument for that compacted bentonite is homogeneously structured, Glaus et al. (2007) still analyze their results from the perspective of the mainstream view, and do not — in my view — fully conclude what their results imply.
In particular they postulate the presence of an interlayer domain and a “free pore water” domain, and write for the “total” flux1 (their eq. 3)
\begin{equation} J_\mathrm{tot} = J_\mathrm{il} + J_\mathrm{pw} \tag{1} \end{equation}
where \(J_\mathrm{il}\) is a presumed diffusive flux in the interlayer domain and \(J_\mathrm{pw}\) is the presumed diffusive flux in the “free pore water” domain.
Their subsequent analysis shows that the measured flux in montmorillonite scales as
\begin{equation} J_\mathrm{tot} \propto \frac{1}{\left ( C_\mathrm{bkg.}\right )^Z} \tag{2} \end{equation}
where \(C_\mathrm{bkg.}\) is the concentration of the background electrolyte (NaClO4), and \(Z\) is the charge number of the diffusing tracer (\(Z = 1\) for sodium and \(Z=2\) for strontium). Moreover, by considering ion exchange equilibrium, Glaus et al. (2007) show that also \(J_\mathrm{il}\) is expected to scale according to eq. 2. As they also confirm that this scaling behavior is not observed in systems without interlayer pores (kaolinite), they could have confidently concluded that their results imply that interlayers are the only significant pore structure in montmorillonite at these densities (as the title suggests).
Unfortunately, the discussion part of the article is considerably more tentative, focusing mainly on “interpretations” of the resulting flux
The present work shows that the interpretation of cation diffusion experiments in highly compacted swelling clays in terms of the concentration gradient in the aqueous phase may result in a nonsensical dependence of the effective diffusion coefficients on the salt concentration in the external aqueous phase. An alternative interpretation using an effective diffusion coefficient in the interlayer water (\(D_\mathrm{il}\)), being independent of the external salt concentration, with a corresponding concentration gradient in the interlayer water is more consistent with the experimental observations.
and the article ends on a quite apologetic note
The proposed interpretation should in turn not be blindly applied to other experimental conditions. Diffusion of cations via the free pore water may become increasingly important in swelling clays with lower degrees of compaction or in clays in which the interlayer gel pores are not that adjacent as they are in compacted montmorillonite. In such cases, the assumption of \(J_\mathrm{tot} \cong J_\mathrm{il}\) may no longer hold, and a double-porous diffusion model would have to be applied in such cases. The present concept may also reach its limits when dealing with cations that rather sorb by surface complexation than by ion exchange. Further work is therefore planned to extend the investigations to such systems.
Given that the mainstream view to this day continues to be the default approach, one may think that this “further work” did show some convincing evidence for e.g. “diffusion of cations via the free pore water” at lower density. But what has actually been shown is that the “assumption of \(J_\mathrm{tot} \cong J_\mathrm{il}\)“ continues to be true for lower density!
Before we look at the additional results, we summarize the findings of Glaus et al. (2007).
Findings in Glaus et al. (2007)
In the following we will consider the so-called “effective diffusion coefficient”, here strictly defined as the experimental parameter
\begin{equation} D_e = -\frac{j_\mathrm{ss}\cdot L}{\Delta c^\mathrm{ext}} \tag{3} \end{equation}
where \(j_\mathrm{ss}\) denotes the steady-state flux when an external tracer concentration difference \(\Delta c^\mathrm{ext}\) is maintained across a bentonite sample of length \(L\). We have discussed through-diffusion and the role of \(D_e\) in many places on the blog, but in the present discussion we simply view \(D_e\) as a normalized version the steady-state flux.
Note that we are required to compare diffusive fluxes in different montmorillonite samples (an alternative test protocol is suggested below). \(D_e\) varies both due to varying background concentration (which is our object of study) and due to the variation of different samples. It is thus crucial to minimize the latter type of variation. This should be done (I suppose) by employing as identical preparation protocols as possible. We will get back to this complication of sorting out signal from noise as we comment the results.
Glaus et al. (2007) present their results in diagrams where the logarithm of the evaluated quantities (diffusion parameters) is plotted against background concentration. This is of course convenient, as e.g. \(D_e\) can be expected to vary by two orders of magnitude as the background concentration is varied between 0.01 M and 1.0 M. But to remind ourselves what the actual dependency looks like between the normalized steady-state flux and background concentration, I will here insist on plotting the results in lin-lin diagrams.
The results for sodium in Glaus et al. (2007) plotted in lin-lin diagrams, look like this (the data is the same in these three diagrams)

We see that the data complies with the scaling law (eq. 2) and is quite well constrained (click on pictures to enlarge). \(D_e\) is evaluated in two ways in Glaus et al. (2007): by examining at the breakthrough curve, and by examining the internal tracer profile at test termination. These methods of evaluation give more or less identical results, with the exception of the test performed at 0.01 M background concentration. In this low concentration limit, the confining filters increasingly restrict the flux, making it difficult to extract actual clay transport parameters. We have discussed this issue (and this particular study) at length in a previous blog post.
Even with the problem of accurately measuring \(D_e\) at the lowest background concentration, the results clearly demonstrate the behavior of a homogeneous system (eq. 2): e.g., \(D_e\) undoubtedly increases by a factor of approximately 10 when the background concentration is lowered from 1.0 M to 0.1 M.
The data for strontium in Glaus et al. (2007) only covers the background concentration interval 0.5 M — 1.0 M, and is consequently less constrained, as seen here

This data also has the peculiarity that the diffusivity of samples of length 5.4 mm is almost twice as large as for samples of length 10.4 mm. This clearly demonstrates how sample preparation becomes crucial when conducting these types of tests. In the plots above, I have allowed myself to treat samples of different length separately (Glaus et al. (2007) use average values). It is clear from the data, that also strontium is compatible with the scaling law of eq. 2. In particular, it can be distinguished that sodium and strontium have different dependencies.
The take away message from these results is clear: montmorillonite at this density (1950 kg/m3) behave as a homogeneous system and shows no indication of containing additional pore structures.
Glaus et al. (2013) and NTB-17-12
After the publication of Glaus et al. (2007), corresponding results for lower densities has been presented. Glaus et al. (2013) — which is mostly recognized for demonstrating the seeming “uphill” diffusion effect — also contains measured \(D_e\) of sodium as a function of background concentration in conventional through-diffusion tests, both for density 1600 kg/m3 and 1300 kg/m3. These results are also published in more detail,2 together with new strontium results, in the NAGRA technical report NTB-17-12. We therefore look at these two publications together.
The additional data for sodium is here compared with the results from Glaus et al. (2007)

For some of the additional tests, both through- and out-diffusion were performed. These points are labelled “TD” and “OD”, respectively, in the diagrams. We see that even for density as low as 1300 kg/m3, the data complies with the behavior of a homogeneous system (eq. 2) and is quite well constrained; in particular, there is nothing in the data for 1300 kg/m3 that suggests that these systems behave principally different than the 1950 kg/m3 samples.
For the system at 1300 kg/m3 and background concentration 0.1 M, two different values of \(D_e\) are presented in NTB-17-12. Only the lower of these values (\(7.0\cdot 10^{-10}\) m2/s) was published in Glaus et al. (2013), but NTB-17-12 presents a continued analysis that includes filter resistance, giving the value of \(D_e\) presented in the diagram. I think this is quite interesting, as the tests made at 0.1 M used “flushed” filters in order to minimize filter resistance. Apparently, filter resistance is still influential and it is not that easy to “design away” this problem.
NTB-17-12 also presents measured values of \(D_e\) for strontium under similar conditions (1300 — 1900 kg/m3, 0.1 — 1.0 M NaClO4 background), and are here compared with the earlier results

Although it naturally contains some scatter, we note that the additional data for ~1900 kg/m3 strengthens the earlier conclusion that also strontium scales in accordance with eq. 2. And just as for sodium, we see that the behavior does not qualitatively change, even for densities as low as 1300 kg/m3.
In the above diagrams are plotted single values for \(D_e\) for strontium at the lowest background concentration (0.1 M). It should be noted that these are burdened with large uncertainties as the transport restriction of the confining filters is severe; in NTB-17-12 are presented a whole set of simulations of the underlying flux evolution and concentration profiles with variations of the filter transport parameters. It is thus very clear that the problem of eliminating transport restrictions at the sample interfaces are not easy to completely eliminate. This is not surprising, as the theory suggests that \(D_e\) increases without limit with decreasing background concentration. Note that this behavior is strongly enhanced for divalent strontium; the measured values are many times larger than the corresponding diffusivity in bulk water (\(0.79\cdot 10^{-9}\) m2/s).
Even if the value of \(D_e\) is quite uncertain at the lowest background concentration, the mere observation that filter diffusivity strongly influence the process is, in a sense, itself a confirmation that the system still is governed by the behavior of interlayers.
The picture is quite clear from these findings: the combined results of Glaus et al. (2007), Glaus et al. (2013) and NTB-17-12 validates a homogeneous view of compacted bentonite, at essentially any relevant density!
The curious case of Bestel et al. (2018)
Bestel et al. (2018) further examine how \(D_e\) for sodium varies with background concentration. This publication shares some of the same authors with the previous studies, and presents additional measurements of \(D_e\) for sodium in essentially identical systems (similar preparation protocols, “Milos” montmorillonite, NaClO4 background electrolyte, flushed filters). Given the substantial evidence for homogeneous behavior collected in the publications discussed above, I find the conclusions of Bestel et al. (2018) rather odd.
Bestel et al. (2018) perform subsequent measurements of the steady-state flux in the same samples at different temperatures. The dependency of \(D_e\) on background concentration, however, looks essentially the same for each temperature, and — just as Bestel et al. (2018) — we here focus mainly on the results for 25 \(^\circ\mathrm{C}\). This data looks like this3

In their analysis, Bestel et al. (2018) include the results from Glaus et al. (2007) and Glaus et al. (2013), but treat them separately. They consequently conclude implicitly that, although the earlier studies found that \(D_e\) depends on background concentration in accordance with eq. 2, the new results show a different behavior. Specifically, they conclude that \(D_e\) scale with background concentration as \(C_\mathrm{bkg}^{-0.52}\) for density 1300 kg/m3 and as \(C_\mathrm{bkg}^{-0.76}\) for density 1600 kg/m3. Bestel et al. (2018) write
The results obtained in the present work for a broad variety of bulk dry densities of Na-montmorillonite and concentrations of the background electrolyte, give clear evidence that the equilibrium distribution of cations between the clay phase and the external aqueous phase is the main parameter influencing the observed overall diffusive fluxes of cations. Whether the observed overall diffusive fluxes are described by a physical subdivision of the pore space into domains containing different species (e.g. the model proposed in Appelo and Wersin (2007) or Bourg et al. (2007)), or whether they are the result of the concentration gradients of such species in a single type of pore (e.g. the model proposed by Birgersson and Karnland (2009)), cannot be decided unambiguously from the available data — notably because of the wide similarity of the model predictions and because of some internal inconsistencies in the experimental data. Both types of models would require some adjustments in order to fully match the data. The diffusion data of \(^{22}\mathrm{Na}^+\) can equally be described by a surface diffusion model with a reduced, but non-zero mobility of sorbed cations, similar to the median value determined in Gimmi and Kosakowski (2011).
I think this is a problematic way of arguing and presenting data.
The data obviously has scatter
To begin with, why are the results from this study and the ones from Glaus et al. (2007) and Glaus et al. (2013) treated separately? When treated separately — according to Bestel et al. (2018) — these results are vaguely supposed to be incompatible: the dependence of \(D_e\) either comply with eq. 2 or it does not. I think that the appropriate thing to do is to discuss possible causes for why the new results supposedly differ from the earlier ones. As we have made clear above, all factors that determine \(D_e\) are not fully controlled in tests like these (e.g, what causes the difference in diffusivity for strontium in 5.4 mm and 10.4 mm samples, respectively, in Glaus et al. (2007)?). We have also seen that it is difficult to make accurate measurements at low enough background concentration, even with flushed filters.
Look e.g. at the specific values of \(D_e\) at background concentrations 1.0 and 0.1 M, respectively, in NTB-17-12 and Bestel et al. (2018) (unit is m2/s).
| Study | 1300 kg/m3 0.1 M | 1300 kg/m3 1.0 M | 1600 kg/m3 0.1 M | 1600 kg/m3 1.0 M |
| NTB-17-12 | \(8.6\cdot 10^{-10}\) | \(0.59\cdot 10^{-1 0}\) \(0.64\cdot 10^{-10}\) | \(3.3\cdot 10^{-10}\) | \(0.43\cdot 10^{-1 0}\) \(0.44 \cdot 10^{-10}\) |
| Bestel et al. (2018)4 | \(4.5\cdot 10^{-10}\) | \(1.2\cdot 10^{-1 0}\) \(1.0\cdot 10^{-10}\) | \(3.4\cdot 10^{-1 0}\) \(3.5\cdot 10^{-10}\) | \(0.53\cdot 10^{-1 0}\) \(0.50\cdot 10^{-10}\) |
Under ideal conditions, these values would not differ for the same conditions in the two studies. The scatter of these values is moreover quite random, e.g. one study do not have values that are systematically larger than in the other. In Bestel et al. (2018) we also see that the mere disturbance of a sample in form of a temperature pulse may alter the diffusivity significantly (temperature is first increased in steps from 25 \(^\circ\mathrm{C}\) to 80 \(^\circ\mathrm{C}\), then decreased in steps to 0 \(^\circ\mathrm{C}\), and finally increased again to 25 \(^\circ\mathrm{C}\)). In e.g. one sample of density 1600 kg/m3 and background concentration 0.1 M is reported \(D_e = 3.4\cdot 10^{-10}\) m2/s at 25 \(^\circ\mathrm{C}\) before the conducted temperature changes, and \(2.3\cdot 10^{-10}\) m2/s after. One should also consider that the samples are not prepared equally, as they are saturated directly with the corresponding background solution. (This is also true for the previous studies.) Could this cause differences in diffusivity?
Bestel et al. (2018) should thus either argue for why the new results are more accurate (or why the results of Glaus et al. (2007, 2013) are less accurate) or treat the data from all studies in accumulation and admit substantial experimental uncertainty. My impression is that Bestel et al. (2018) make a little of both.
The data still complies with a homogeneous view
Looking at the aggregated sodium data, a somewhat different picture emerges

Here is also included a model labelled “Full Donnan”, which takes into account the excess salt that is expected to enter the interlayers. For all other samples we have discussed, this contribution is only minor and can be neglected, and this assumption underlies eq. 2. For the sample of density 1300 kg/m3 with background concentration 5.0 M, however, the excess salt is not negligible and must be included in the analysis of the behavior of a homogeneous system (the deviation from eq. 2 is seen to become significant around 1.0 M background concentration). Bestel et al. (2018) actually present a full Donnan calculation for the excess salt, but, for unknown reasons, do not compare it directly with the experimental results (it is plotted in a separate diagram next to the data).
For 1300 kg/m3, I would claim that the “Full Donnan” model fits better to the accumulated data than the scaling law suggested in Bestel et al. (2018) (exponent \(-0.52\)). For 1600 kg/m3, the suggested scaling law (exponent \(-0.76\)) indeed fits better to the data than eq. 2, but the data is not that well constrained. To use this singular result to argue for a non-homogeneous bentonite structure basically boils down to claiming that the values measured at 0.1 M — a concentration range that is documented to be difficult to measure accurately — could not possibly be underestimated by, say, 50% (while also ignoring all other results).
If we also consider the results for strontium presented in NTB-17-12, I mean that the only reasonable conclusion that Bestel et al. (2018) can draw is that the results comply with a homogeneous bentonite structure.
Additional model components should not be motivated solely by the ability of a model to be fitted to some arbitrary data
A major motivation for measuring how \(D_e\) depends on background concentration at lower densities, according to Glaus (2007), is that “the assumption of \(J_\mathrm{tot} \cong J_\mathrm{il}\) may no longer hold”. What (I mean) has been demonstrated in the subsequent studies is that this assumption actually does hold. In particular, from the aggregated data it is not possible to claim that the behavior of \(D_e\) is qualitatively different at 1950 kg/m3 and 1300 kg/m3. Thus, there is no valid justification for introducing more complex model components. Moreover, introducing e.g. a bulk water phase causes fundamental conflicts with the description of other well-established properties of these systems, particularly swelling pressure. Adding such components merely to improve agreement with a specific dataset, while ignoring their broader implications, undermines the model’s overall coherence and validity. The data cannot “equally be described by a surface diffusion model”.
What does some alternative model actually predict?
Eq. 2 (or a full Donnan calculation) is a clean statement of the expected behavior of a homogeneous system (based on how interlayers function). If actual deviations from this behavior could be established we may conclude that a homogeneous description is not sufficient. However, any arbitrary deviation from eq. 2 does not automatically validate any specific alternative model. Validating a model requires that we can experimentally reproduce some of its non-trivial predictions. Bestel et al. (2018) don’t discuss what the exponents \(-0.52\) and \(-0.76\) are suppose to represent.
Note also that the arbitrary exponent \(-0.52\) is inferred by fitting to the data at 5.0 M background concentration. But we saw above that a full Donnan calculation within the homogeneous view actually explains the behavior in this concentration limit (Bestel et al. (2018) show this!). We have thus every reason to believe that the exponent \(-0.52\) is just spurious and do not represent some actual physical mechanisms.
We should also keep in mind that any reasonable validity of the model of Gimmi and Kosakowski (2011) has not been demonstrated, not even by the data supplied in that particular publication. I have written a separate blog post on this issue.
A suggestion for how to preferably conduct these types of tests
The discussed studies are enough to convince me that cation tracer diffusion behave in accordance with a homogeneous bentonite view at any relevant density.
It is however also clear that the full variation of \(D_e\) in these tests is caused by more factors than just background concentration and density. To eliminate as much as possible of this scatter — and thus to more accurately determine the dependence of background concentration on \(D_e\) — I suggest the following test protocol.
- Measure tracer flux at several background concentrations in the same sample.
This would eliminate both the unavoidable (small) variation in density between different samples as well as several unknown factors that determine the exact value of diffusivity (these may e.g. be related to variation in material or equipment and to sample handling) - Prepare samples by saturating them all with the same low concentration solution (e.g. 0.05 M).
To me it seems reasonable that the way samples saturate may influence the resulting detailed structure and thus the diffusivity. Saturating all samples in the same manner with the same solution will minimize variations from such effects.
- Keep temperature constant.
I don’t think this is a crucial factor, but we see in Bestel et al. (2018) that larger temperature pulses may significantly alter the diffusivity. - Increase background concentration in steps and record the steady-state flux at each concentration.
I think a good range may be between 0.2 M and 1.0 M. For a homogeneous system, this corresponds to a variation in \(D_e\) by a factor 5 for monovalent and 25 for divalent cations.5 At the same time, the problem of filter transport resistance can hopefully be kept under control. - Decrease the background concentration (perhaps in steps) back to the first concentration where steady-state flux was measured.
Measure steady-state flux again and assert that no significant change in \(D_e\) has occurred as a consequence of the disturbance introduced by the background concentration pulse.
Final thoughts
The only reasonable conclusion to draw from the studies we have looked at is that the behavior of cation tracer diffusion indicates a relatively homogeneous structure, dominated by interlayers, in any relevant bentonite system. Despite this, the contemporary scientific bentonite literature is crammed with non-homogeneous descriptions of compacted bentonite, centered around a bulk water phase (the “mainstream view”). As we have seen here, this can even be the case for studies that provide evidence for homogeneity.
What I find most frustrating is that interlayer effects often are viewed as some additional feature to be handled in specific cases. In reality, virtually all experimental findings (diffusion, swelling pressure, temperature response, Donnan effects, fluid flow, hyperfiltration, …) indicate that the behavior of compacted bentonite is fully governed by interlayers. The question is not if a presumed bulk water phase may dominate under certain conditions, but if such a phase is at all relevant. I want to emphasize this point: up until this day, no convincing evidence has ever been presented that compacted bentonite contains significant amounts of bulk water.
Even if the structure becomes more complex at lower densities, a homogeneous model centered around interlayers guarantees to cover at least some aspects of the system. On the contrary — if the goal is process understanding — most experimental evidence rules out bentonite models that assume a bulk water phase.
Footnotes
[1] We here ignore that diffusive fluxes are not additive.
[2] As far as I can see, these tests were done in duplicates for Na diffusion with background concentrations 0.5 M and 1.0 M, and the the numbers reported in Glaus et al. (2013) are averages.
[3] Bestel et al. (2018) use a normalization scheme in their analysis that involves corresponding measured water diffusivities and parameters from “Archie’s law” (note, it is the quotation marks version of the law). I think this handling makes the presented results less transparent, and here we use the actual reported values of \(D_e\).
[4] These are only values from the first phase at 25 \(^\circ\mathrm{C}\).
[5] I assume that measurements are being made in pure Na-montmorillonite.






































{kind=link}
{kind=link}