
In the ongoing assessment of chloride equilibrium concentrations in bentonite, we here take a closer look at the study by Glaus et al. (2010), in the following referred to as Gl10. We thus assess the 4 points indicated here

Reading Gl10 gives the impression that the study consists solely of through-diffusion tests of a set of different tracers (HTO, sodium, chloride), in a set of different materials (Kaolinite, “Na-Illite”, Na-montmorillonite), at nominal density 1.9 g/cm3. A lot of additional information, however, is published in a later, completely separate publication: Glaus et al. (2011), which we will refer to as Gl11. Needless to say, this is a quite peculiar way of reporting a study. For instance, Gl10 do not provide any geometrical information about the samples (!), but this is found in Gl11; Gl11 also report corresponding out-diffusion measurements that apparently were made.1
Even with the combined sources of Gl10 and Gl11, information is not entirely complete. For example, tests have been carried out in duplicates, but evaluated diffusion parameters are only reported as averages (table 2 in Gl10). Furthermore, the sources give contradictory information in some instances (this is further discussed below). Scraping both sources for information, these are the tests that have been performed, as far as I understand:
- Through-diffusion
In total 8 separate tests were performed, with NaClO4 background concentrations of 0.1 M, 0.5 M, 1.0 M and 2.0 M. These were performed in sequence in four different tests cells. Thus, two tests at 1.0 M background concentration were first performed in two different samples; thereafter, the same two samples were used for two additional tests at 2.0 M. Similarly, in two other samples, two 0.5 M tests were followed by two 0.1 M tests. The steady-state concentration profile in the clay was measured in one single test, performed at 0.1 M background concentration.In this assessment we will also make use of the results from through-diffusion of water (HTO). These were made at background concentrations 0.1 M and 1.0 M. We will return to the question of whether they were carried out in the same samples as used for the chloride-diffusion experiments.
- Out-diffusion
Most of the through-diffusion tests were followed by out-diffusion tests: after steady-state was reached, the external reservoirs were exchanged for tracer free solutions, and diffusion of chloride out of the sample was recorded.Out-diffusion was tested on all samples at background concentrations 0.5 M and 2.0 M, and on one sample at background concentration 0.1 M.
- Sorption
The montmorillonite material was tested for sorption of chloride, in suspensions with background concentrations of either perchlorate or chloride (at 0.5 M). - Equilibrium tests
At least one test was conducted to investigate the amount of ClO4 in the clay after the sample was equilibrated with a specified external concentration. - Investigation of swelling during dismantling.
The samples were cylindrical with diameter 2.54 cm, and with slightly different lengths, close to 1.0 cm. The sample volume is thus roughly 5 cm3.
In the following, we mainly refer to the chloride diffusion tests in montmorillonite. Although the diffusion parameters are only reported as averages, each individual parameter is actually found in a single plot in Gl10 (“Fig. 6”). From this plot we can extract results from each individual through-diffusion test (see below).
In Gl10 are also presented breakthrough curves (flux vs. time) for four tests, one for each different background concentration. Similarly, in Gl11 are presented three flux-vs.-time plots for out-diffusion. As will be further discussed below, we have to do some combined guess- and detective work in order to identify these flux evolution curves with specific samples.
Material
The material is referred to as montmorillonite “from Milos”, and was prepared specifically for the study. Bentonite from Milos (Greece), purchased from Süd-Chemie (now Clariant), was repeatedly washed in strong NaCl solutions to remove most of the accessory minerals and to convert the clay to essentially pure sodium-form. Excess NaCl was subsequently removed from the clay by dialysis. Gl10 present analyses of the chemical composition of both the used materials, as well as of a further purified 0.5 \(\mu\)m fraction of the montmorillonite material. From these analyses it is concluded that the used montmorillonite still contains some silica accessory minerals (3 — 4%), as well as some carbonate (calcite). We may thus assume a montmorillonite content of around 95%.
Concerning the cation population, Gl10 assert that the detected calcium is “most probably” present as CaCO3 rather than being part of the exchangeable cations. However, as the purification procedure used here is quite similar to that used in Muurinen et al. (2004) — that we have assessed earlier — we may expect some influence of calcium on the exchangeable cations. Muurinen et al. (2004) measured a Na/Ca-ratio of approximately 90/10 in their material, which also contained some carbonate (as well as sulfate). Here we assume that the used Na-montmorillonite is basically a pure sodium system, but should keep in mind that the presence of calcium may somewhat influence the results, especially since the different samples are exposed to very different external sodium concentrations.
Sample density
The nominal density for all samples appears to be 1.9 g/cm3, but actual sample densities are not reported (in Gl10, it is even hard to find information on nominal density). However, results of HTO diffusion in four tests (at 0.1 M and 1.0 M background concentration) indicate a considerably lower density. Porosities inferred from the breakthrough curves for these tests range between approximately 0.35 — 0.42. As is further discussed below, we here choose a range for the porosity of 0.321 — 0.394. Assuming a grain density of \(\rho_s\) = 2.8 g/cm3, this corresponds to a density range of 1.9 g/cm3 — 1.7 g/cm3 (effective montmorillonite density 1.87 g/cm3 — 1.66 g/cm3).
Uncertainty of external solutions
We have no reason to doubt the validity of the solutions used, and will assume no uncertainty here.
Evaluations from the diffusion tests
The chloride diffusion data in Gl10 and Gl11 is essentially analyzed in terms of the effective porosity model, although the fitted parameters are the “effective diffusivity” (\(D_e\)) and the “rock capacity factor” (\(\alpha\)). But for chloride, Gl10 use \(\alpha\) and \(\epsilon_\mathrm{eff}\) (the “effective porosity”) interchangeably.2 To avoid confusion, we will only use the notation \(\epsilon_\mathrm{eff}\).
As mentioned, Gl10 only tabulate the mean values of \(D_e\) and \(\epsilon_\mathrm{eff}\) for each background concentration, but we can extract each individual parameter graphically. The extracted \(D_e\) and \(\epsilon_\mathrm{eff}\) are listed here.3
| \(C_\mathrm{bkg}\) M | \(\epsilon_\mathrm{eff}\) (Gl10 fig. 6) | \(D_e\) 10-13 m2/s (Gl10 fig. 6) | \(D_p\) 10-12 m2/s eq. 1 | \(D_p\) 10-12 m2/s from sim. | \(j_\mathrm{ss}\) 10-13 mol/m2/s from sim. | \(c^\mathrm{source}\) mol/m3 eq. 2 |
| 0.1 | 0.017 | 0.68 | 4.0 | 3.9/3.4 | 1.51/1.67 | 0.021 |
| 0.1 | 0.021 | 0.75 | 3.6 | 4.3 (profile) | 1.8 (in text) | 0.024 (in text) |
| 0.5 | 0.036 | 2.5 | 6.8 | 6.6 | 5.46 | 0.022 |
| 0.5 | 0.036 | 2.5 | 6.8 | 6.3 | 11.7 | 0.046 |
| 1.0 | 0.050 | 4.9 | 9.8 | 10 | 10.9 | 0.023 |
| 1.0 | 0.050 | 4.6 | 9.2 | |||
| 2.0 | 0.068 | 7.4 | 11 | 10.6/10.5 | 16.3/16.8 | 0.022 |
| 2.0 | 0.066 | 6.9 | 10 |
With a single exception, the averages are identical with what is listed in table 2 in Gl10, which confirms the accuracy of the extracted parameters (for 1.0 M background concentration, the average \(\epsilon_\mathrm{eff}\) is 0.050 rather than the tabulated value 0.051). In the above table are also listed the corresponding pore diffusivities, evaluated as
\begin{equation} D_p = \frac{D_e}{\epsilon_\mathrm{eff}}. \tag{1} \end{equation}
From the flux and profile data found in Gl10 and Gl11, we can also evaluate several pore diffusivites ourselves. Such values are presented in the fifth column in the above table, and corresponding steady-state fluxes are found in the sixth column. Below is compared various flux vs. time data with my own simulations.



Regarding the breakthrough curves, the test design is here much better than what we have encountered in earlier assessments; the transient stage is properly sampled rather than that the data mainly represents a sequence of steady-state measurements.4 This makes the inference of diffusion parameters quite easy and robust.
Comparing the through-diffusion and out-diffusion results we can conclude that the data presented in Gl10 and Gl11 for background concentration 0.1 M most probably is for the same sample. Although the fitted parameters differ somewhat, the text of Gl11 states a steady state flux of 1.8⋅10-13 mol/s/m2 for the other 0.1 M sample, which was subsequently sectioned. As the presented through-diffusion flux is considerably smaller we may conclude that this is the same sample for which out-diffusion subsequently was conducted.
For the 0.5 M data, we can instead conclude that the two data sets must stem from two different samples, as the steady-state fluxes differ by roughly a factor of 2. For the 2.0 M data, the fitted parameters are very similar for the two test phases, which may indicate that they were measured in the same sample. However, the parameters are also very similar for the other test. The same is true for 1.0 M data (for which no out-diffusion was performed).
From steady-state fluxes and reported values of \(D_e\), we can calculate the corresponding tracer concentration in the source reservoir as
\begin{equation} c^\mathrm{source} = \frac{j_\mathrm{ss}\cdot L}{D_e} \tag{2} \end{equation}
where \(L\) is sample length.5 Source tracer concentrations evaluated in this way are presented in the last column in the above table (source concentration is only reported for a single test, in Gl11).
Finally, we can also look at the presented tracer profile at termination, which was determined in a single case,6 for one of the 0.1 M tests.

We note — as does Gl11 — that the concentration profile shows quite extensive interface excess, a topic that we have discussed in a separate blog post. The main focus of Gl11 is actually a modeling treatment of these regions, but here we focus on the linear interior part of the profile.7 Fitting a line to this part (see figure) we extract a slope of -22.0 nmol/g/m. Gl11 do not report the corresponding density profile (that most certainly was measured), but using the nominal density (1.9 g/cm3), gives a corresponding clay concentration gradient of \(\nabla c_\mathrm{ss} = -0.0418\) mol/m4. Combining this value with the steady-state flux (1.8⋅10-13 mol/m2/s; reported in the text in Gl11), we can independently evaluate the pore diffusivity
\begin{equation} D_p = -\frac {j_\mathrm{ss}}{\nabla c_\mathrm{ss}} = \frac{1.8\cdot 10^{-13}}{0.0418} \;\mathrm{m^2/s} = 4.3 \cdot 10^{-12} \;\mathrm{m^2/s} \end{equation}
This is in reasonable agreement with the value evaluated from \(D_e\) and \(\epsilon_\mathrm{eff}\).
In conclusion, even though crucial information is missing in Gl10, the re-evaluations made here, with help from information in Gl11, confirm the adequacy of the reported parameters \(D_e\) and \(\epsilon_\mathrm{eff}\). A perhaps single conspicuous detail is that the source concentration in one of the 0.5 M tests appears to have been about twice as large as for any of the other tests. There may, of course, be a reasonable explanation for this.
Evaluating chloride equilibrium concentrations
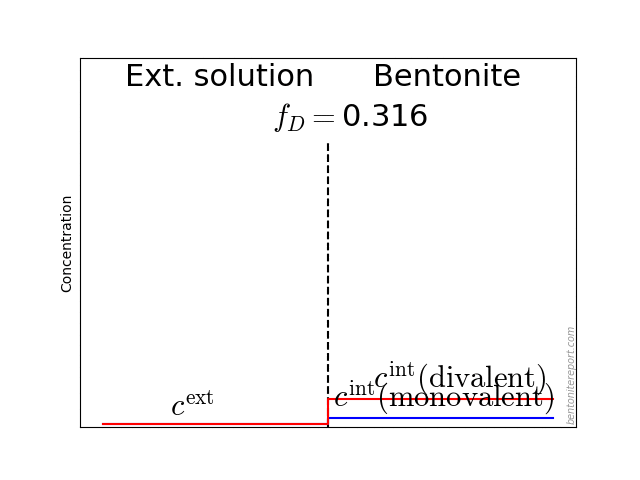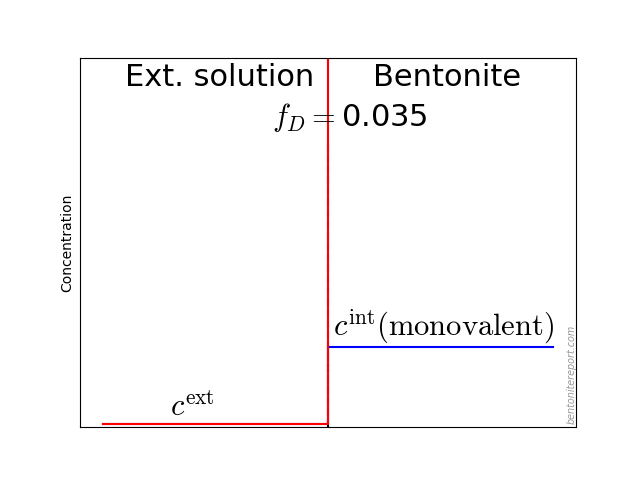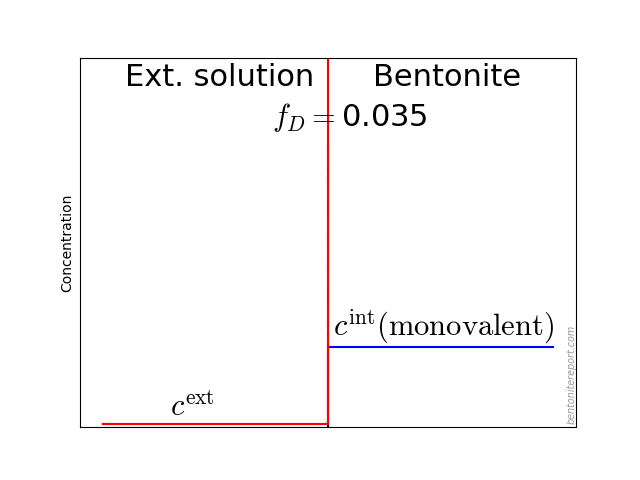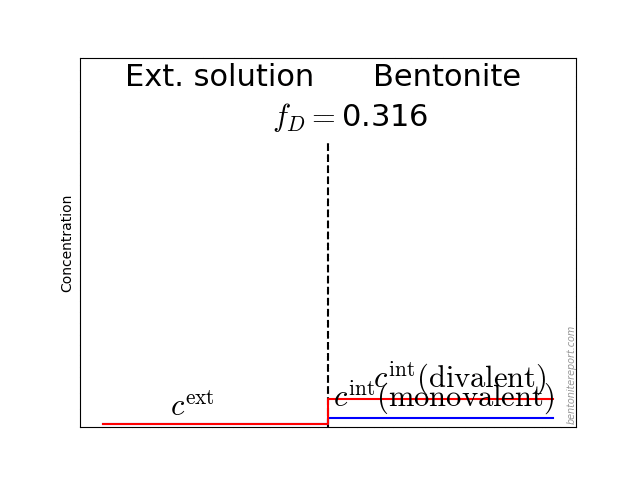
As noted in earlier assessments, the convenient quantity expressing the chloride equilibrium in through-diffusion tests is the ratio \(\bar{c}(0) / c^\mathrm{source}\), where \(\bar{c}(0)\) denotes the tracer concentration within the clay, at the interface to the source reservoir (for details, see here).
From the reported values of \(\epsilon_\mathrm{eff}\), the most straightforward way to evaluate the chloride equilibrium concentrations is
\begin{equation} \frac{\bar{c}(0)}{c^\mathrm{source}} = \frac{\epsilon_\mathrm{eff}}{\phi} \tag{3} \end{equation}
where \(\phi\) is the (physical) porosity. Gl10 (or Gl11) don’t provide information on actual measured densities, leaving us little choice but to use the nominal density in order to get a value for \(\phi\) in eq. 3. However, Gl10 also provide data for corresponding water (HTO) diffusion measurements. As mentioned above, these measurements indicate densities significantly lower than the nominal value. The (graphically extracted) values for \(D_e\) and \(\epsilon_\mathrm{eff}\) for HTO are
| \(C_\mathrm{bkg}\) M | \(\epsilon_\mathrm{eff}\) (Gl10 fig. 6) | \(D_e\) m2/s (Gl10 fig. 6) | \(D_p\) m2/s eq. 1 | \(\rho_d\) g/cm3 |
| 0.1 | 0.352 | 1.7e-11 | 4.8e-11 | 1.81 |
| 0.1 | 0.402 | 1.7e-11 | 4.1e-11 | 1.67 |
| 1.0 | 0.401 | 2.0e-11 | 5.0e-11 | 1.68 |
| 1.0 | 0.421 | 1.9e-11 | 4.5e-11 | 1.62 |
For water, the effective porosity parameter is really an estimate of the physical porosity, and we can thus use this value to calculate a corresponding density, which is presented in the last column in the table.
Gl10 state
The diffusion of the various radioactive tracers (HTO, 22Na, 36Cl) was measured in sequence, each new tracer run was started after the out-diffusion of the previous tracer had been completed.
which is hard to interpret in any other way than that the above HTO parameters have been evaluated in the same samples in which chloride diffusion was tested. However, the protocol presented in Gl11 does not include any HTO diffusion “measured in sequence” (see above for information on the test protocol). The two sources evidently contain some contradictory information.8 Under any circumstance, as water diffusivity is claimed to be measured in samples with the same nominal density, we must assume a quite substantial uncertainty of the actual sample densities. In evaluating the chloride equilibrium concentrations, we therefore choose a porosity interval between the nominal value and the average given from the water parameters: \(\phi\sim\) 0.321 — 0.394. The table below lists the corresponding intervals for the chloride equilibrium concentrations
| \(C_\mathrm{bkg}\) M | \(\epsilon_\mathrm{eff}\) (Gl10 fig. 6) | \(\bar{c}(0)/c^\mathrm{source}\) eq. 3 | \(\bar{c}(0)/c^\mathrm{source}\) outflux (eq. 4) / profile |
| 0.1 | 0.017 | 0.043 — 0.053 | 0.053 — 0.065 (o) |
| 0.1 | 0.021 | 0.053 — 0.065 | 0.051 — 0.070 (p) |
| 0.5 | 0.036 | 0.091 — 0.112 | |
| 0.5 | 0.036 | 0.091 — 0.112 | 0.107 — 0.131 (o) |
| 1.0 | 0.050 | 0.127 — 0.156 | |
| 1.0 | 0.050 | 0.127 — 0.156 | |
| 2.0 | 0.068 | 0.173 — 0.212 | 0.170 — 0.213 (o) |
| 2.0 | 0.066 | 0.169 — 0.206 |
From the out-diffusion tests we can also evaluate the equilibrium concentrations “independently”, by integrating the flux. As discussed in the assessment of Van Loon et al. (2007), this integral (multiplied by sample area) gives one third of the total amount of tracers present in the clay at the start of the out-diffusion phase (these quantities are labelled “Acc.” in the above diagrams). With an estimate of the tracer concentration in the source reservoir, the equilibrium chloride concentration can thus be evaluated as
\begin{equation} \frac{\bar{c}(0)}{c^\mathrm{source}} = \frac{6\cdot N_\mathrm{right}}{\phi \cdot V_\mathrm{sample} \cdot c^\mathrm{source}} \tag{4} \end{equation}
where \(N_\mathrm{right}\) denotes the final amount of tracers in the target reservoir. The corresponding chloride equilibrium concentrations are listed in the last column in the above table.
Finally, we also look at the 0.1 M test for which the steady-state tracer concentration profile was recorded. Extrapolating the linear part to the clay/source interface, gives a chloride content of 0.282 nmol/g, which corresponds to a clay concentration interval of 5.37⋅10-4 — 4.80⋅10-4 mol/m3, using the porosity interval defined above.9 Given the source concentration (0.024 mol/m3), these values correspond to a chloride equilibrium concentration ratio in the range 0.051 — 0.071.
The different ways of estimating chloride equilibrium concentrations provide a quite consistent picture (see above table). Although the information has been difficult to extract, it may thus seem that, in the end, all is good and well. However, we should note that the evaluated pore diffusivities show a quite peculiar dependency on background concentration.

Such a dependency, which has not been observed in earlier assessed studies, directly influence the evaluated equilibrium concentrations. As the breakthrough curves are so well sampled in the present study, this result can hardly be attributed to uncertainty in the values of \(D_p\). While Gl10 don’t explicitly identify this behavior (they do not evaluate \(D_p\)), a main focus of the study is actually to account for it, by means of “Archie’s law”, i.e. by suggesting a non-linear functional relationship between \(D_e\) and \(\epsilon_\mathrm{eff}\). I am strongly critical of such a treatment, but will refrain from discussing it here, as the focus of this assessment is the data itself rather than its interpretation (we have discussed this issue in a previous blog post).
An obvious alternative interpretation of this behavior is that chloride adsorbs on some system component, in the sense of becoming immobilized (what I have earlier dubbed true sorption). Gl11 test this hypothesis by performing additional batch sorption tests on the montmorillonite, in background solutions of NaCl and NaClO4 (0.5 M) at various pH. Although they cannot exclude a “\(R_d\)” value of the order of 10-4 m3/kg, they ultimately conclude that chloride do not sorb to any significant extent in these systems (and continues with “explaining” the behavior as resulting from other mechanisms).
I mean, however, that some experimental observations suggest that a sorption mechanism may be active. In addition to the above limit for the \(“R_d”\) value, we may note significant chloride sorption in the kaolinite samples, which were also studied in Gl10. There may of course be a reasonable explanation for why chloride sorption is observed in kaolinite, while it is not active in montmorillonite, but this issue is not really discussed in Gl10. Also, the recorded steady-state chloride content profile suggests a non-zero value at the interface to the target reservoir. This could, reasonably, indicate that some chloride is immobilized.
Perchlorate equilibrum concentrations
On the other hand, an additional argument against chloride sorption is that equilibrium perchlorate concentrations seem to be comparable with those evaluated for chloride. Gl11 don’t report perchlorate content directly, and we have to do some work to extract the corresponding equilibrium concentration in the 0.1 M sample that was sectioned. Gl11 plot the chloride tracer content for this sample together with “the concentration in the anion-accessible volume”, labelled \(c_\mathrm{acc}\).

\(c_\mathrm{acc}\) is, unsurprisingly, not a directly measured chloride concentration, but a quite elaborate interpretation of the data. From the unreported ClO4 content, an “anion-accessible porosity” variable has been calculated, by simply multiplying the physical porosity by the ratio between internal and external ClO4 concentrations. \(c_\mathrm{acc}\) is, in turn, defined as the actual measured chloride content distributed in a volume that corresponds to this “anion-accessible porosity”. By combining the reported chloride content (let’s call it \(\bar{n}_\mathrm{Cl}\)) and \(c_\mathrm{acc}\), we can thus de-derive the perchlorate equilibrium concentration as
\begin{equation} \frac{\bar{c}_\mathrm{ClO_4}}{0.1 \;\mathrm{M}} = \frac{\bar{n}_\mathrm{Cl}\cdot\rho}{c_\mathrm{acc}\cdot \phi} \end{equation}
Using this formula for the inner “linear” part of the profile (2 — 8 mm) gives the values 0.060, 0.059, 0.061 and 0.062, assuming nominal density. For porosity 0.394 the corresponding values are 0.044, 0.043, 0.044, and 0.045. We note that a range 0.043 — 0.062 for the equilibrium concentration ratio at 0.1 M background is in line with the previous evaluations. It should be noted, though, that this evaluation is for perchlorate, which not necessarily has the same equilibrium concentration as chloride. Nonetheless, this evaluation shows a similar, relatively high, equilibrium concentration also for this ion.
In fact, Gl11 provide results from yet another test where the focus is the perchlorate equilibrium,10 this time at a background concentration of 0.5 M. The results are reported as physical and “anion-accessible” porosities, evaluated from measuring water and perchlorate content.11

We note that also this sample shows substantial interface excess, but here we focus on the inner, relatively flat part (marked points in figure). From values of physical and effective porosity, we can directly calculate an equilibrium concentration in accordance with eq. 3. In this case the equilibrium concentration can also be related to a measured density. Using the average values gives a perchlorate equilibrium concentration ratio of \(\bar{c}_\mathrm{ClO_4}/0.5\; \mathrm{M} = 0.150\). Note that this value should be associated with density of 2.05 g/cm3 (the average porosity for the inner points is 0.259). This perchlorate equilibrium concentration ratio is nevertheless considerably larger than what was evaluated for chloride at (nominal) density 1.9 g/cm3 (0.11). This may indicate that perchlorate has a larger preference for the clay than chloride in these systems, but, as 2.05 g/cm3 is remarkably high, I suspect that measured water contents in this test have been systematically underestimated.
Summary and verdict
With only the information given in Gl10, I would judge the provided information too uncertain to be used for quantitative process understanding of chloride equilibrium in bentonite. With the additional information provided in Gl11, however, we have seen that the diffusion parameters — and consequently the equlibrium concentrations that can be inferred — can be assessed to have been quite robustly evaluated. Needless to say, access to a completely separate publication should not be needed in order to make this type of assessment. Nevertheless, my choice is to keep this data to use for evaluating e.g. performance of models for salt exclusion.
A remaining uncertainty is the actual density of the tested samples. Results from corresponding water tracer tests suggest densities considerably lower than the nominal density. It not fully clear, however, if these water diffusion tests were conducted with separate samples or with the same samples as for the chloride diffusion tests.
Finally, these results complicate the picture of chloride equilibrium concentrations in bentonite, as they do not fully comply with earlier ones. In particular, here is observed a dependency of the pore diffusivity on the background concentration, and chloride contents, which are not seen in other studies. For anyone that is truly interested in how salts distribute in bentonite, it should be a priority to understand how the present results can be reconciled with other chloride equilibirum results.12
Below is plotted the chloride equilibrium concentrations evaluated from this study. For each background concentration is drawn an “uncertainty box”, that takes into account the uncertainty in density, as discussed above, and the corresponding interval in equlilibrium concentration ratio. The corresponding points have been arbitrarily put in the middle of these “uncertainty boxes”. The effective montmorillonite density has been calculated assuming a montmorillonite content of 95%.

To compare the present results with others, we have also plotted some chloride equilibrium concentration evaluated from Van Loon et al. (2007), that we have assessed previously.
- Chloride content: UNKNOWN
- Extracting anion equilibrium concentrations from through-diffusion tests
- Assessment of chloride equilibrium concentrations: Muurinen et al. (1988)
- Assessment of chloride equilibrium concentrations: Molera et al. (2003)
- How salt equilibrium concentrations may be overestimated
- Assessment of chloride equilibrium concentrations: Muurinen et al. (2004)
- Assessment of chloride equilibrium concentrations: Van Loon et al. (2007)
- Assessment of chloride equilibrium concentrations: Muurinen et al. (2007)
- Assessment of chloride equilibrium concentrations: Ishidera et al. (2008)
Footnotes
[1] To be fair, reading Gl10 carefully, out-diffusion is briefly mentioned a couple of times.
[2] Gl10 rather use the term “accessible porosity”, and symbol \(\epsilon_\mathrm{acc}\), but we stick with the terminology that we have used in the previous assessments. Also, a critique of mixing the effective porosity model (that involves \(\epsilon_\mathrm{eff}\)) and the traditional diffusion-sorption model (that involves \(\alpha\)) is found here.
[3] For background concentration 0.5 M it is difficult to resolve if the diagram in Gl10 has a single point, or if there are two points on top of each other. As Gl10 claim that duplicates were made at all concentrations, here we have assumed two different samples with identical parameters.
[4] The through-diffusion flux evolution for background concentration 0.1 M plotted in Gl10 seems not to be complete: the diagram shows data points up until day 160, but Gl11 state that the test was conducted for 229 days.
[5] The simulations presented here use \(L\) = 9.75 mm for the samples with background concentration 2.0 M, and \(L\) = 10.25 mm for the samples with background concentrations 0.1 M and 0.5 M. These are average values from the sample lenghts reported in Gl11.
[6] In Gl10 is stated that
Tracer profiles of 36Cl in Na–mom were found to be in qualitative agreement with those found by Molera et al. (2003) and exhibited two distinct linear regions with different slopes. In contrast to Molera et al. (2003) we interpret the 36Cl profiles in terms of heterogeneities of compaction in the boundary zones of the clays and not as the result of two diffusion processes. In view of these ambiguities, tracer profiles were generally used as a consistency test and not for the calculation of \(D_e\) values.
At least to me, this way of writing gives the impression that profiles were recorded for most of the tests. In Gl11, however, we learn that only a single profile was recorded.
[7] Gl11 argue for that the non-linear parts of the profile actually reflect the state of the sample during steady-state, rather than being an effect of dismantling. I am strongly critical to their arguments, and plan to comment on this in a separate blog post.
[8] For the sodium measurements in montmorillonite, it is certain that the above statement is false. Most of these were made in 5.4 mm samples, and they were all sectioned. Morover, these were reported in a much earlier publication: Glaus et al. (2007).
[9] The clay concentration is calculated as \(\bar{c} = \bar{n} \cdot \rho_d/\phi\), where \(\bar{n}\) denotes the chloride concentration as amount per dry mass.
[10] The main focus in Gl11 is actually the density distribution in the interface regions of the sample, but this is a straightforward perchlorate equilibrium test.
[11] The data in this plot has been “de-scaled”, as it was measured in a 5.4 mm sample, but then “recalculated” (!?) for a 10 mm sample in Gl11.
[12] I intend to write a follow-up blog post discussing these issues.






















{kind=link}