Over a long period on the blog, we have systematically examined studies on chloride equilibrium in sodium dominated bentonite. We have now individually assessed each study that was deemed to have potential to provide relevant information. In this blog post we make some overall conclusions and give an updated picture of what is actually known empirically regarding chloride equilibrium in bentonite.
The assessment included seven studies, which are summarized in the table below. The table also provides links to each individual assessment.
These studies are the only ones, to my knowledge, that meet the
following criteria:
They involve chloride
There are both theoretical and empirical arguments for that different anions may have different equilibrium concentrations (for otherwise similar conditions). In the assessment it has therefore been important to stick to one and the same type of equilibrating anion. Moreover, chloride is certainly the anion that has been studied the most within bentonite research, with iodide as its closest “competitor”.
They involve sodium dominated bentonite
This include commercial products, such as “MX-80”, “Kunigel V1” or “Kunipia F”, or materials that were intentionally prepared for the study (more or less pure Na-montorillonite).
Some studies exist where ion equilibrium is explored in other systems, e.g. claystone or bentonites dominated by divalent counter-ions. But, since we have every reason to belive that the conditions for ion equilbrium are different in such systems, as compared to Na-bentonite, we must be careful not to include them in the analysis. We shouldn’t compare apples and oranges.
They have a specified external sodium solution
Without some knowledge of the composition of the solution in contact with the sample, an evaluated chloride concentration cannot be related to any relevant equilibrium condition. Furthermore, if the water chemistry of the equilibrating solution is too complex (e.g. involving several cations), the equilibrium cannot in a reasonably straghtforward manner be related to chloride concentrations in a sodium dominated system.
They have a systematic variation of either density or external background concentration or both
My main motivation for making these assessments is for using equilibrium data to better understand salt exclusion in bentonite. This can reasonably only be achieved if density and/or background concentration has been systematically varied.
In the following we will refer to each study with the identifying
label listed in the table above.
Comments
Through-diffusion is unneccesary
A majority of the examined studies are
through-diffusion studies (Mu88, Mo03, Vl07, Is08, Gl10). A
through-diffusion test set-up is, in fact, much more complex than
required for only studying equilibrium quantities: it involves
monitoring the chemical evolution of the external solutions (often
using radiochemical methods), and the final state (steady-state)
concentration profile is often extracted, by meticulously sectioning
and analyzing the sample (studies where final state profiles were
extracted are indicated by a “p” in the above table).
Additionally, extracting relevant information from flux data requires fitting a two-parameter model. In all assessed diffusion studies, one parameter relates to mobility (either an “effective” or an “apparent” diffusion coefficient) and one to ion equilibrium (“effective porosity”, “anion-accessible porosity”, or a “capacity factor”).1 Consequently, through-diffusion tests, despite their complexity, only provide indirect estimates of equilibrium concentrations, and the accuracy of the estimated parameters naturally depends on details of the fitting procedure and the sampled data. In this regard, most of the studies we have examined report inferior fitting procedures and flux data, where the transient stage of the process has not been adequately sampled (the only exception being Gl10).2 Estimated “effective porosities” are therefore not very reliable. This imprecision can sometimes be mitigated by also using information on the final state concentration profile. But this part of the analysis then essentially corresponds to making a quite complicated equilibrium test. Two of the five diffusion studies — Mu88 and Mo03 — were discarded because evaluated parameters (and the underlying data) are too uncertain.
From an ion equilibrium perspective, through-diffusion tests are
consequently not very “economical”.
The obvious alternative are straightforward equilibrium
tests, where samples simply are equilibrated with specified external
solutions. This can in principle be done without monitoring, and only
requires the patience to wait long enough. The lack of any requirement
to monitor these types of tests also makes them suitable, I imagine,
for involving many samples without significantly increasing the
experimental workload.
Most equilibrium tests have not been adequately performed
Although they are conceptually much simpler, only two of the assessed
studies are pure equilibrium tests (Mu04 and Mu07).
A third (Vl07) performed explicit equilibrium measurements
as part of a diffusion study.
Essentially all studies in the assessment that have recorded concentration profiles show interface excess, i.e. an increased amount of ions near the edges as compared with the interior of the samples. As this effect seems to be universal,3 it must be accounted for when making equilibrium tests, or evaluated concentrations will be overestimated. Doing this should be quite straightforward, by, for example, quickly sectioning off the first few millimeters on both sides of the samples during dismantling. Unfortunately, this has not been done in the assessed equilibrium studies,4 which makes them unsuitable. Vl07, on the other hand, recorded full profiles, and the excess effect was accounted for.
Relevant parameter ranges
After discarding two diffusion studies and two equilibrium studies,
only three studies remain for which the evaluated equilibrium
concentrations are deemed sufficiently accurate: Vl07, Is08 and Gl10.
But we should also consider the relevance of the chosen density and background concentration ranges — something that has not been discussed to any greater extent in the individual assessments. My main motivation for performing this assessment is for using equilibrium concentration data for testing models for salt exclusion in compacted bentonite. A full understanding of ion equilibrium in such systems is crucial for e.g. a relevant chemical description of bentonite buffers in radioactive waste repositories. Therefore, a preferred effective montmorillonite density range is approximately 1.2 — 1.7 g/cm3, say.
With also this criteria in mind, we may therefore rule out two of the three remaining studies; Is08 treats low density systems (\(<\) 1.0 g/cm3), and Gl10 only considers an extremely high high density (1.9 g/cm3).5 This leaves us with a single study that passes both the test of providing accurate data on chloride equilibrium concentrations and being measured in relevant parameter ranges: Vl07. This study covers the approximate density range 1.15 — 1.75 g/cm3, and concentration range 0.01 — 1.0 M.
A single relevant study
On the one hand, it is great news that we have verified some data as
actually useful for evaluating salt exclusion in compacted
bentonite. On the other hand, it is very unfortunate that there only
is one single study!
Moreover, although the results of Vl07 most definitely are useful, they are not optimal. A more “pragmatic” problem with this study is that it reports whole sets of “Cl-accessible porosities”1 for each sample tested, together with an average value. But these different values simply reflect the uncertainty of the parameter for individual samples. If the study had no issues (experimental or modeling related), these values should all be the same, as they are evaluated from one and the same sample. In our assessment we identified that the major part of this uncertainty stems from evaluations from diffusion modeling, while estimations made from equilibrium considerations are more robust (total out-diffusion and stable chloride content). It is thus these estimations in Vl07 that are deemed useful, while the diffusion estimations should be discarded. Note that, since “Cl-accessible porosities” estimated from flux data are sub-optimal, so are the reported average values.
Unfortunately,
severalstudies have
used or
reported
the Vl07 data (as well as other data we have assessed) without
sufficient rigor when evaluating salt exclusion in compacted
bentonite. As a relatively recent example of this,
Gimmi and Alt-Epping (2018) compare two models for chloride exclusion with empirical data
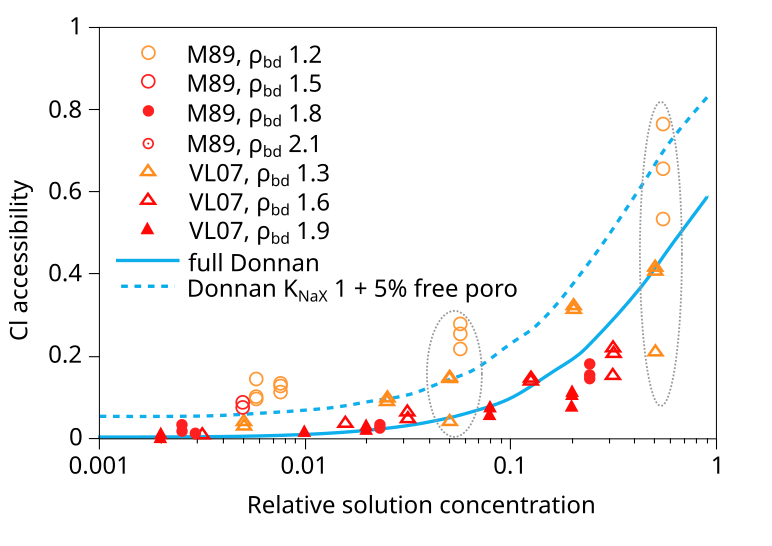
in a figure that looks very similar to this6
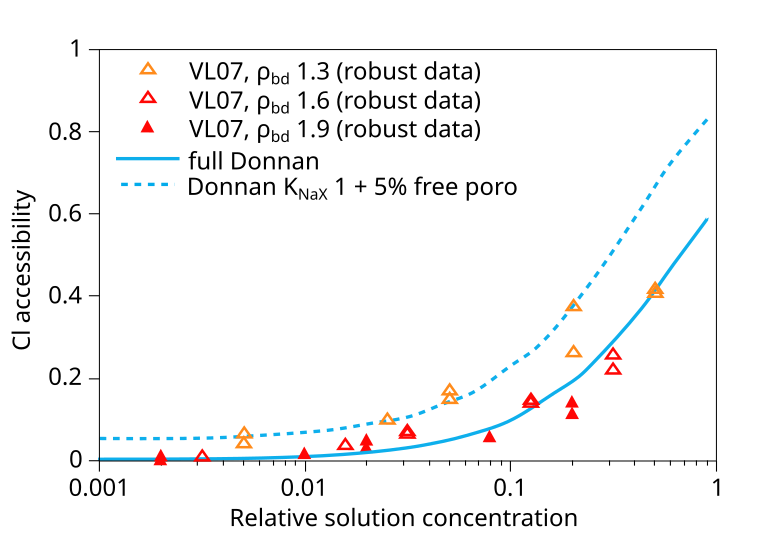
In addition to the VL07 data, this plot also compare with data from Mu886 — a study that we have discarded. Taking the above plot at face value it is hard not to wonder what use the experimental data really has — the spread at certain places is almost an order of magnitude (indicated in the figure). You can basically fit any favorite model to this data (or rather, you can fit no model to this data). Gimmi and Alt-Epping (2018) anyway compare the data with two Donnan equilibrium models. One (“full Donnan”) is essentially equivalent to the homogeneous mixture model (all pore space is treated equally), while the other includes several specific additional model components (“free” porosity, exchange “sites”). Gimmi and Alt-Epping (2018) use this plot to argue for that these particular additional components become significant for bentonite at the lower density. But if we “clean up” the plot and only use data points that has passed the present assessment, the picture is instead this
With this version of the data we can at least convince ourselves that
it obeys the rules for Donnan equilibrium. But I mean that it is hard
to draw any more detailed conclusions than that. In particular, it is
a hard stretch to believe that the suggested more complex model has
any particular significance.7
The Vl07 data also has the more fundamental problem that the detailed
ionic composition of the system is not fully controlled. This is
actually the case for all assessed studies that use “natural”
bentonite rather than specifically prepared homoionic clay, and
relates to to the presence of uncontrolled amounts of divalent
cations.
Problems with ignoring the detailed equilibrium conditions
When “natural” bentonites — which generally contain more than one
type of cation — are contacted with a pure sodium solution, it is
inevitable that the material and the solution begin exchanging
cations. Furthermore, since these materials contain accessory
minerals, dissolution/precipitation processes are most probably also
initiated. Thus, at the time when the equilibrium concentration is
recorded, the exact chemical conditions are typically not known. In
particular, it is not clear exactly what e.g. the Na/Ca ratio is in
the clay. To make issues worse, the extent of this effect depends
significantly on the concentration of the external solution, where we
expect a purer sodium clay for higher external concentrations. Since
the external concentration is often varied by orders of magnitude in
these studies, this implies that quanities evaluated at different
concentrations most likely correspond to slightly different systems
(e.g. clay samples with different Na/Ca ratios). Thus, even if we have
taken measures when selecting studies to not compare apples and
oranges, this problem partly remains.
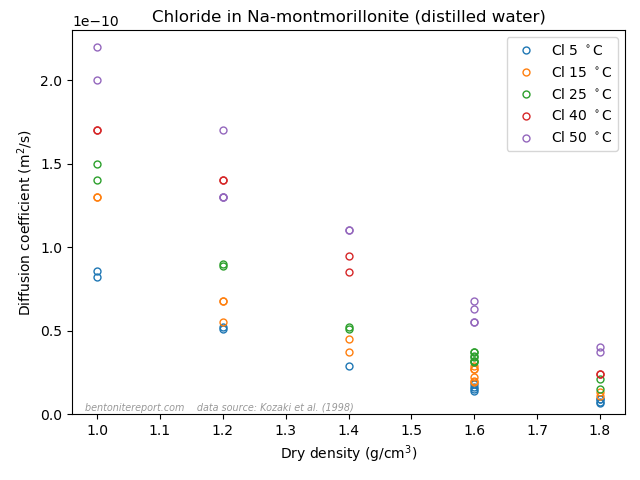
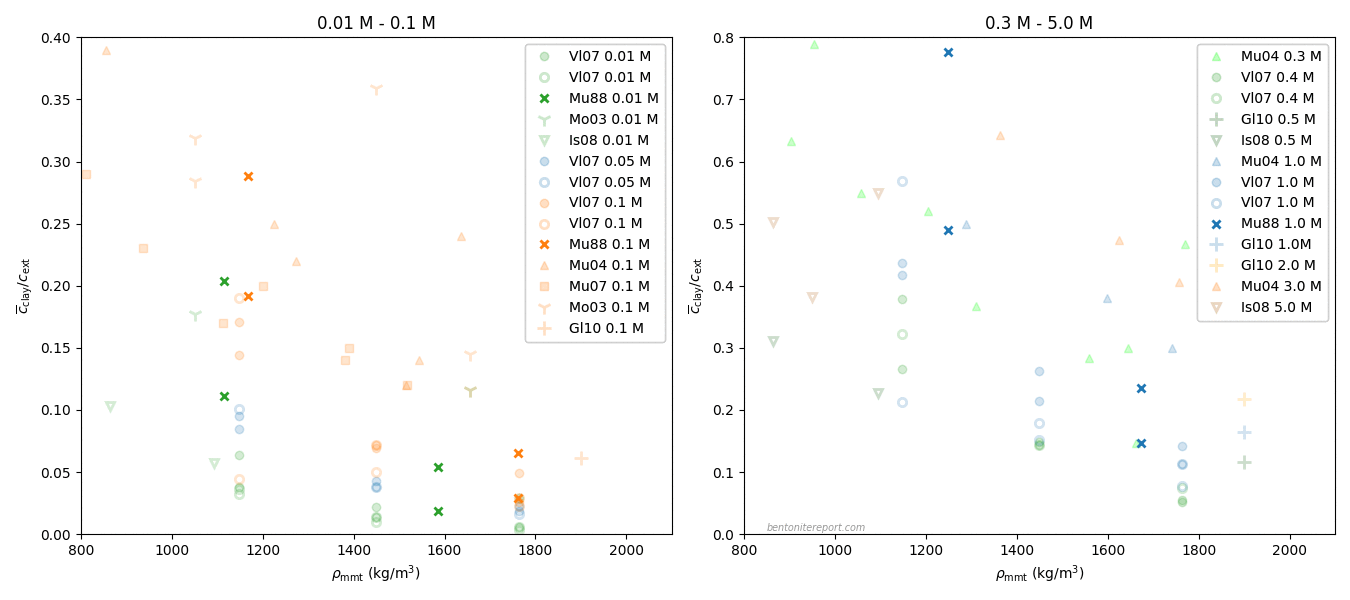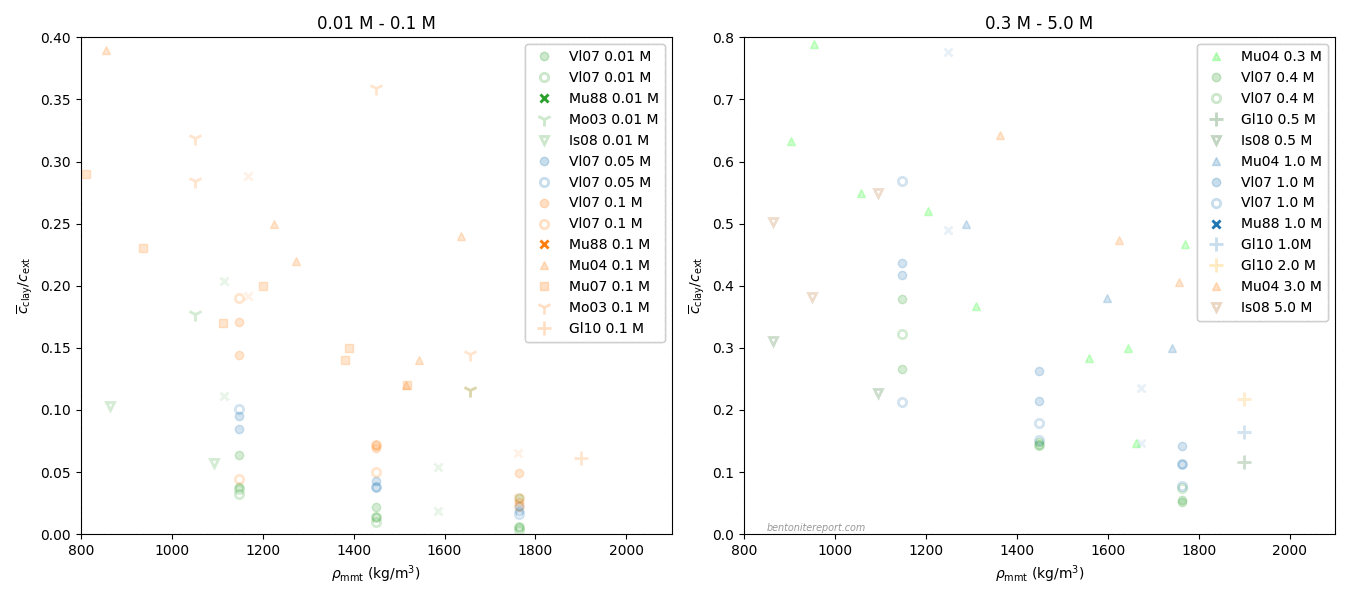
Relevant data for chloride equilibrium concentrations
Below is plotted the chloride equilibrium data that has been found
robust and relevant in the assessment (i.e. part of the data reported
in Vl07)
These values have been evaluated from the “Cl-accessible porosities” reported in table 6 in Vl07.8 The exact values of equilibrium concentration ratios and effective montmorillonite densities depend on adopted values for grain density and montmorillonite content. Here we have adopted \(\rho_s\) = 2800 kg/m3 and 80% montmorillonite. Note that equilibrium concentrations and densities are burdened with additional uncertainties that are not indicated in the above diagram. Note also that although most conditions in the above plot have two data points, these correspond to a single sample. For more details we refer to the individual assessment.
Comparing the above plot with
the one presented in the initial blog post on the assessment —
which included all available data — we note a considerably
less chaotic picture. At least, the robust Vl07 data gives evidence
for the two main features that we discussed in the initial blog post:
Chloride exclusion increases with increasing density at constant background concentration
Chloride exclusion decreases with increasing background concentration at constant density
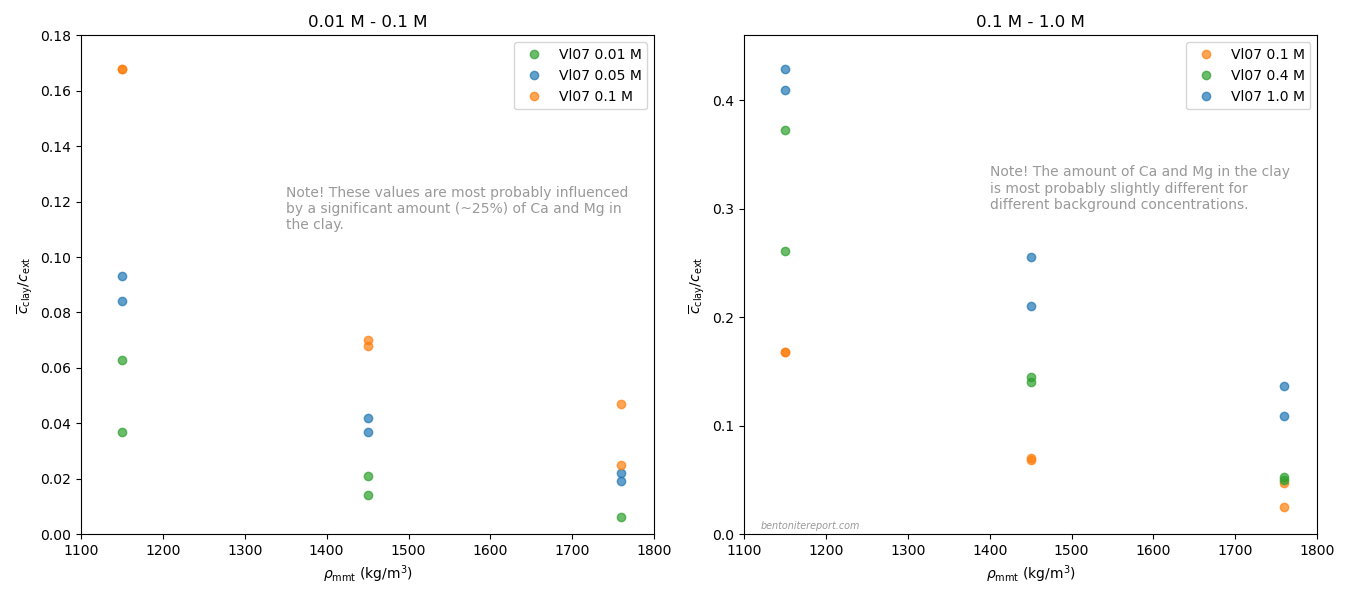
It must be emphasized that the Vl07 data most probably has a
systematic “error”, in the sense that the data for lower background
concentrations (0.01 — 0.1 M) most probably is influenced by a
significant amount of divalent exchangable cations in the clay (Ca and
Mg). In contrast, for higher background concentrations (0.4 M, 1.0 M),
the clay is most probably in a purer sodium state.
A hundred labs should each make a hundred equilibrium tests!
After finishing this assessment the loudest question in my head is: why are not a hundred labs already on their way to each make a hundred equilibrium tests? Not only has the bentonite research sector failed when we must rely on a single soon 20-year-old study to have some idea of chloride equilibrium in sodium dominated bentonite. For other anions we essentially have no systematic data! As mentioned above, a general understanding of ion equilibrium is required in order to perform relevant chemical modeling of e.g. bentonite buffers in radioactive waste repositories.
[1] Here we do not discuss the
reasonability of these models and model parameters. I am, however,
arguing heavily in many
otherplaces on the blog that none of them are conceptually
sound. Here I have described how experimentally accessible equilibrium
concentrations can be extracted from “anion-accessible porosity”
parameters.
[2] This bad test design
isstillverycommon.
Through-diffusion tests should reasonably be designed so that the
outflux curve can be adequately sampled. As this curve behaves
drastically differently in the transient and in the steady-state
stages, the sampling frequency should reasonably be adapted.
As an example, if a lab has the capacity to make measurements at most every second day (as is done in e.g. Vl07), I suggest starting diffusion tests on a Friday and design them so that essentially no tracers reaches the target reservoir during the weekend. This can be achieved by aiming for a breakthrough time of about 20 days. The breakthrough time is related to diffusivity (\(D\)) and sample length (\(L\)) as \begin{equation*} t_\mathrm{bt} = \frac{L^2}{6D} \end{equation*} Consequently, to keep \(t_\mathrm{bt}\) relatively constant, sample lengths should be adjusted depending on the expected value of the diffusivity. For a breakthrough time of 20 days, \(D = 10^{-10}\) m2/s corresponds to \(L=32\) mm, and \(D = 10^{-11}\) m2/s to \(L=10\) mm.
With a breakthrough time of about 20 days, and tests started on
Fridays, I suggest the following measurement protocol
3 times a week the first 3 weeks (Monday, Wednesday, Friday)
2 times a week the following 3 weeks (Monday, Friday)
1 time a week the following 5 weeks (Friday)
This would give sampling at 20 occasions over about 80 days that
ideally corresponds to four times the breakthrough time, like this
However, I further argue for that through-diffusion tests generally
should be avoided. Diffusivities are more conveniently (and quickly)
measured in closed-cell tests. Likewise, for equilibrium properties
it is obviously better to perform equilibrium
tests. Through-diffusion tests, in my opinion, are only motivated
under
particular circumstances, e.g. for making several non-destructive
measurements in the same sample under various conditions.
[3] I am fully convinced that this is an effect due to swelling during sample dismantling. Molera et al. (2003) and Glaus et al. (2011) have presented other interpretations, which we have briefly discussed in the assessments. I intend to write a future separate blog post on this topic.
[4] Sample
information in Mu07 is sparse and it is not clear how dismantling
has been performed, but nothing suggests that interface excess has
neither been identified nor handled. In the individual assessment of
this study I came to the conclusion that this data after all can be
useful for evaluating models for salt exclusion. Here I anyway
discard the results, mainly due to the above mentioned lack of
information. This data should be kept in mind, however.
[5] Both Is08 and Gl10 provide interesting information,
which should not be completely forgotten. In particular, Is08 report
results for extremely high background concentrations (5.0 M). Gl10,
on the other hand, show a dependency on background concentration of
the diffusivity not seen in other tests. I was not able to rule out
this effect as an artifact and therefore encourage the bentonite
research community to help clarify what is occurring in these
specific systems.
[6] The reference for
the points labeled “M89” in
Gimmi and Alt-Epping (2018) is Muurinen et al. (1988), i.e. Mu88. I have not changed the label,
however, because the plot contains more data than what is reported
in Mu88. I have not been able to identify the source for this
additional data. We may also note that the Vl07 data reported here
appears to be quite randomly chosen; for some systems are chosen
data evaluated from diffusion, for others, data evaluated from
equilibrium measurements.
[7] On the contrary, there are many additional arguments for that sodium bentonite at 1.3 g/cm3 does not contain significant amounts of “free” porosity. Moreover, in my head, the procedure of treating ion exchange with both a Donnan equilibrium model and a surface site sorption model can only lead to overparameterization problems. It is also unreasonable in this context to add conceptually completely different features before the “full Donnan” model is treated in full, e.g. by including activity corrections.
[8] One entry in that table, for stable chloride at 1.9 g/cm3 and background concentration 0.01 M, has been discarded. The table also appears to contain a couple of typos, which have been corrected.
Reading Gl10 gives the impression that the study consists solely of through-diffusion tests of a set of different tracers (HTO, sodium, chloride), in a set of different materials (Kaolinite, “Na-Illite”, Na-montmorillonite), at nominal density 1.9 g/cm3. A lot of additional information, however, is published in a later, completely separate publication: Glaus et al. (2011), which we will refer to as Gl11. Needless to say, this is a quite peculiar way of reporting a study. For instance, Gl10 do not provide any geometrical information about the samples (!), but this is found in Gl11; Gl11 also report corresponding out-diffusion measurements that apparently were made.1
Even with the combined sources of Gl10 and Gl11, information is not
entirely complete. For example, tests have been carried out in
duplicates, but evaluated diffusion parameters are only reported as
averages (table 2 in Gl10). Furthermore, the sources give
contradictory information in some instances (this is further discussed
below). Scraping both sources for information, these are the tests
that have been performed, as far as I understand:
Through-diffusion
In total 8 separate tests were performed, with NaClO4 background concentrations of 0.1 M, 0.5 M, 1.0 M and 2.0 M. These were performed in sequence in four different tests cells. Thus, two tests at 1.0 M background concentration were first performed in two different samples; thereafter, the same two samples were used for two additional tests at 2.0 M. Similarly, in two other samples, two 0.5 M tests were followed by two 0.1 M tests. The steady-state concentration profile in the clay was measured in one single test, performed at 0.1 M background concentration.
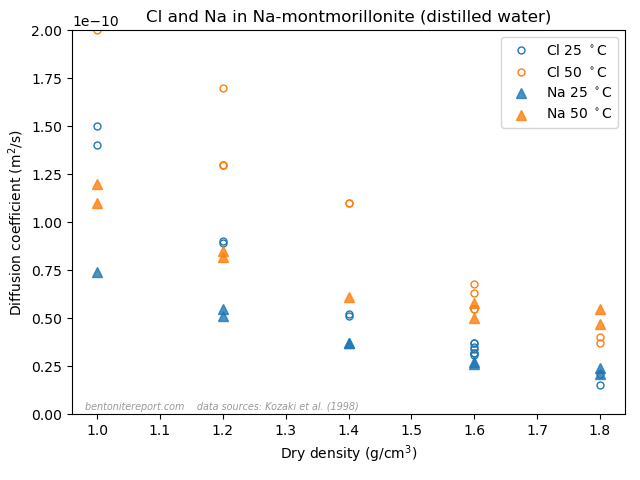
In this assessment we will also make use of the results from through-diffusion of water (HTO). These were made at background concentrations 0.1 M and 1.0 M. We will return to the question of whether they were carried out in the same samples as used for the chloride-diffusion experiments.
Out-diffusion
Most of the through-diffusion tests were followed by out-diffusion tests: after steady-state was reached, the external reservoirs were exchanged for tracer free solutions, and diffusion of chloride out of the sample was recorded.
Out-diffusion was tested on all samples at background concentrations 0.5 M and 2.0 M, and on one sample at background concentration 0.1 M.
Sorption
The montmorillonite material was tested for sorption of chloride, in suspensions with background concentrations of either perchlorate or chloride (at 0.5 M).
Equilibrium tests
At least one test was conducted to investigate the amount of ClO4 in the clay after the sample was equilibrated with a specified external concentration.
Investigation of swelling during dismantling.
The samples were cylindrical with diameter 2.54 cm, and with slightly different lengths, close to 1.0 cm. The sample volume is thus roughly 5 cm3.
In the following, we mainly refer to the chloride diffusion tests in
montmorillonite. Although the diffusion parameters are only reported
as averages, each individual parameter is actually found in a single
plot in Gl10 (“Fig. 6”). From this plot we can extract results from
each individual through-diffusion test (see below).
In Gl10 are also presented breakthrough curves (flux vs. time) for four tests, one for each different background concentration. Similarly, in Gl11 are presented three flux-vs.-time plots for out-diffusion. As will be further discussed below, we have to do some combined guess- and detective work in order to identify these flux evolution curves with specific samples.
Material
The material is referred to as montmorillonite “from Milos”, and was prepared specifically for the study. Bentonite from Milos (Greece), purchased from Süd-Chemie (now Clariant), was repeatedly washed in strong NaCl solutions to remove most of the accessory minerals and to convert the clay to essentially pure sodium-form. Excess NaCl was subsequently removed from the clay by dialysis. Gl10 present analyses of the chemical composition of both the used materials, as well as of a further purified 0.5 \(\mu\)m fraction of the montmorillonite material. From these analyses it is concluded that the used montmorillonite still contains some silica accessory minerals (3 — 4%), as well as some carbonate (calcite). We may thus assume a montmorillonite content of around 95%.
Concerning the cation population, Gl10 assert that the detected calcium is “most probably” present as CaCO3 rather than being part of the exchangeable cations. However, as the purification procedure used here is quite similar to that used in Muurinen et al. (2004) — that we have assessed earlier — we may expect some influence of calcium on the exchangeable cations. Muurinen et al. (2004) measured a Na/Ca-ratio of approximately 90/10 in their material, which also contained some carbonate (as well as sulfate). Here we assume that the used Na-montmorillonite is basically a pure sodium system, but should keep in mind that the presence of calcium may somewhat influence the results, especially since the different samples are exposed to very different external sodium concentrations.
Sample density
The nominal density for all samples appears to be 1.9 g/cm3, but actual sample densities are not reported (in Gl10, it is even hard to find information on nominal density). However, results of HTO diffusion in four tests (at 0.1 M and 1.0 M background concentration) indicate a considerably lower density. Porosities inferred from the breakthrough curves for these tests range between approximately 0.35 — 0.42. As is further discussed below, we here choose a range for the porosity of 0.321 — 0.394. Assuming a grain density of \(\rho_s\) = 2.8 g/cm3, this corresponds to a density range of 1.9 g/cm3 — 1.7 g/cm3 (effective montmorillonite density 1.87 g/cm3 — 1.66 g/cm3).
Uncertainty of external solutions
We have no reason to doubt the validity of the solutions used, and
will assume no uncertainty here.
Evaluations from the diffusion tests
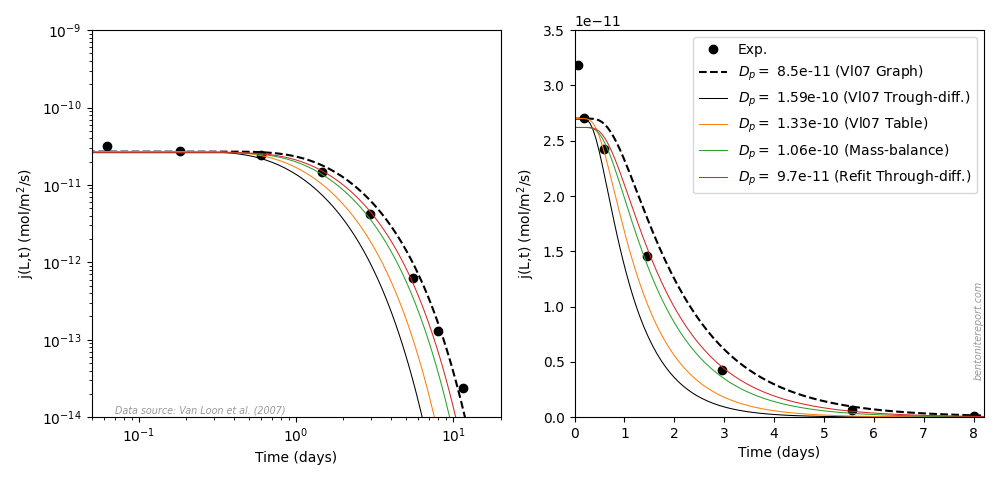
The chloride diffusion data in Gl10 and Gl11 is essentially analyzed in terms of the effective porosity model, although the fitted parameters are the “effective diffusivity” (\(D_e\)) and the “rock capacity factor” (\(\alpha\)). But for chloride, Gl10 use \(\alpha\) and \(\epsilon_\mathrm{eff}\) (the “effective porosity”) interchangeably.2 To avoid confusion, we will only use the notation \(\epsilon_\mathrm{eff}\).
As mentioned, Gl10 only tabulate the mean values of \(D_e\) and \(\epsilon_\mathrm{eff}\) for each background concentration, but we can extract each individual parameter graphically. The extracted \(D_e\) and \(\epsilon_\mathrm{eff}\) are listed here.3
With a single exception, the averages are identical with what is listed in table 2 in Gl10, which confirms the accuracy of the extracted parameters (for 1.0 M background concentration, the average \(\epsilon_\mathrm{eff}\) is 0.050 rather than the tabulated value 0.051). In the above table are also listed the corresponding pore diffusivities, evaluated as
From the flux and profile data found in Gl10 and Gl11, we can also
evaluate several pore diffusivites ourselves. Such values are
presented in the fifth column in the above table, and corresponding
steady-state fluxes are found in the sixth column. Below is compared
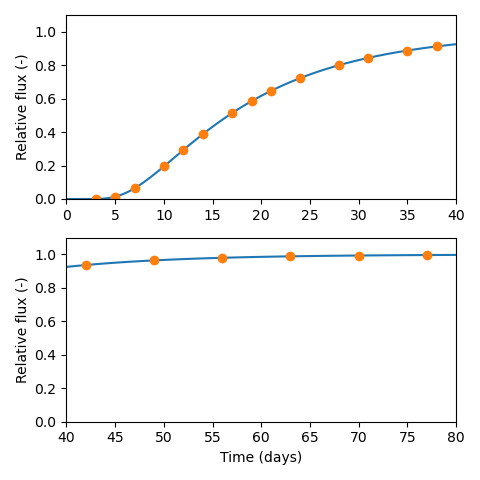
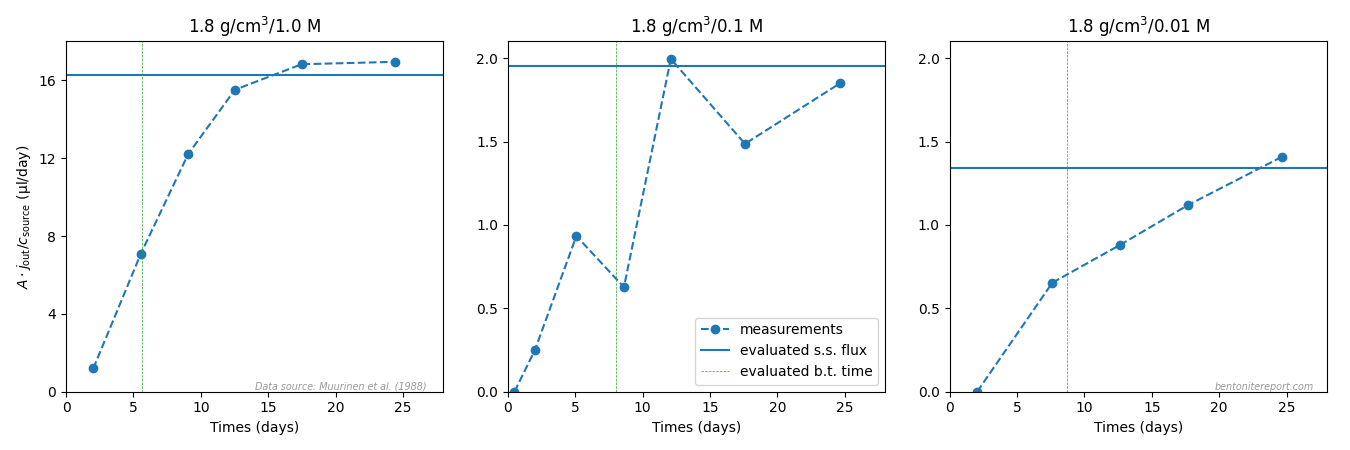
various flux vs. time data with my own simulations.
Regarding the breakthrough curves, the test design is here much better
than what we have encountered in earlier assessments; the transient
stage is properly sampled rather than that the data mainly represents
a sequence of steady-state measurements.4 This makes the inference of diffusion parameters
quite easy and robust.
Comparing the through-diffusion and out-diffusion results we can conclude that the data presented in Gl10 and Gl11 for background concentration 0.1 M most probably is for the same sample. Although the fitted parameters differ somewhat, the text of Gl11 states a steady state flux of 1.8⋅10-13 mol/s/m2 for the other 0.1 M sample, which was subsequently sectioned. As the presented through-diffusion flux is considerably smaller we may conclude that this is the same sample for which out-diffusion subsequently was conducted.
For the 0.5 M data, we can instead conclude that the two data sets must stem from two different samples, as the steady-state fluxes differ by roughly a factor of 2. For the 2.0 M data, the fitted parameters are very similar for the two test phases, which may indicate that they were measured in the same sample. However, the parameters are also very similar for the other test. The same is true for 1.0 M data (for which no out-diffusion was performed).
From steady-state fluxes and reported values of \(D_e\), we can calculate the corresponding tracer concentration in the source reservoir as
where \(L\) is sample length.5 Source tracer concentrations evaluated in this
way are presented in the last column in the above table (source
concentration is only reported for a single test, in Gl11).
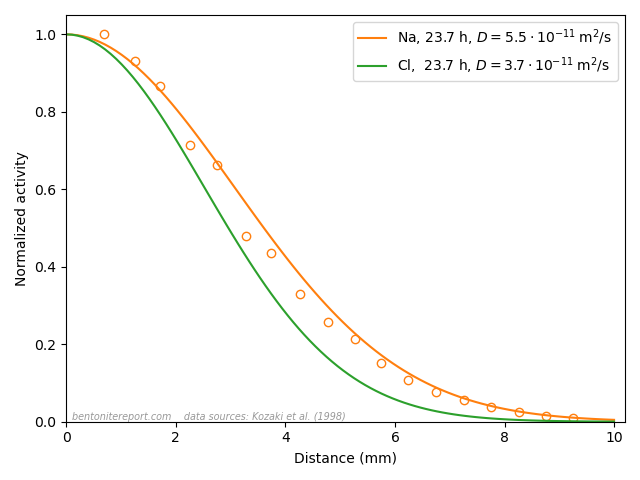
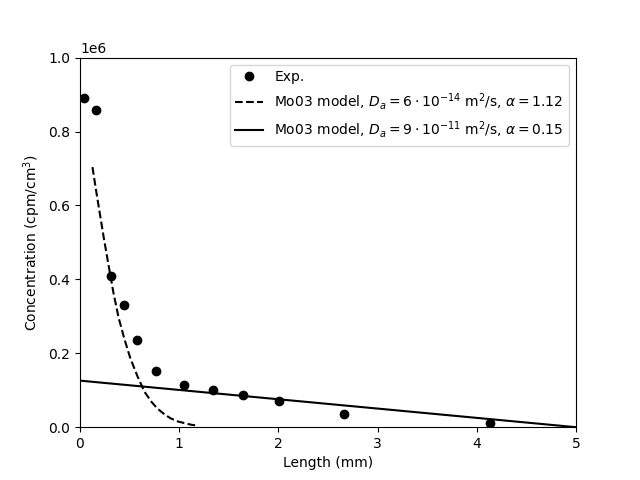
Finally, we can also look at the presented tracer profile at
termination, which was determined in a single case,6 for one of the 0.1 M tests.
We note — as does Gl11 — that the concentration profile shows quite extensive interface excess, a topic that we have discussed in a separate blog post. The main focus of Gl11 is actually a modeling treatment of these regions, but here we focus on the linear interior part of the profile.7 Fitting a line to this part (see figure) we extract a slope of -22.0 nmol/g/m. Gl11 do not report the corresponding density profile (that most certainly was measured), but using the nominal density (1.9 g/cm3), gives a corresponding clay concentration gradient of \(\nabla c_\mathrm{ss} = -0.0418\) mol/m4. Combining this value with the steady-state flux (1.8⋅10-13 mol/m2/s; reported in the text in Gl11), we can independently evaluate the pore diffusivity
This is in reasonable agreement with the value evaluated from \(D_e\) and \(\epsilon_\mathrm{eff}\).
In conclusion, even though crucial information is missing in Gl10, the re-evaluations made here, with help from information in Gl11, confirm the adequacy of the reported parameters \(D_e\) and \(\epsilon_\mathrm{eff}\). A perhaps single conspicuous detail is that the source concentration in one of the 0.5 M tests appears to have been about twice as large as for any of the other tests. There may, of course, be a reasonable explanation for this.
Evaluating chloride equilibrium concentrations
As noted in earlier assessments, the convenient quantity expressing the chloride equilibrium in through-diffusion tests is the ratio \(\bar{c}(0) / c^\mathrm{source}\), where \(\bar{c}(0)\) denotes the tracer concentration within the clay, at the interface to the source reservoir (for details, see here).
From the reported values of \(\epsilon_\mathrm{eff}\), the most
straightforward way to evaluate the chloride equilibrium
concentrations is
where \(\phi\) is the (physical) porosity. Gl10 (or Gl11) don’t provide information on actual measured densities, leaving us little choice but to use the nominal density in order to get a value for \(\phi\) in eq. 3. However, Gl10 also provide data for corresponding water (HTO) diffusion measurements. As mentioned above, these measurements indicate densities significantly lower than the nominal value. The (graphically extracted) values for \(D_e\) and \(\epsilon_\mathrm{eff}\) for HTO are
For water, the effective porosity parameter is really an estimate of
the physical porosity, and we can thus use this value to calculate a
corresponding density, which is presented in the last column in the
table.
Gl10 state
The diffusion of the various radioactive tracers (HTO, 22Na, 36Cl) was measured in sequence, each new tracer run was started after the out-diffusion of the previous tracer had been completed.
which is hard to interpret in any other way than that the above HTO parameters have been evaluated in the same samples in which chloride diffusion was tested. However, the protocol presented in Gl11 does not include any HTO diffusion “measured in sequence” (see above for information on the test protocol). The two sources evidently contain some contradictory information.8 Under any circumstance, as water diffusivity is claimed to be measured in samples with the same nominal density, we must assume a quite substantial uncertainty of the actual sample densities. In evaluating the chloride equilibrium concentrations, we therefore choose a porosity interval between the nominal value and the average given from the water parameters: \(\phi\sim\) 0.321 — 0.394. The table below lists the corresponding intervals for the chloride equilibrium concentrations
From the out-diffusion tests we can also evaluate the equilibrium concentrations “independently”, by integrating the flux. As discussed in the assessment of Van Loon et al. (2007), this integral (multiplied by sample area) gives one third of the total amount of tracers present in the clay at the start of the out-diffusion phase (these quantities are labelled “Acc.” in the above diagrams). With an estimate of the tracer concentration in the source reservoir, the equilibrium chloride concentration can thus be evaluated as
where \(N_\mathrm{right}\) denotes the final amount of tracers in the target reservoir. The corresponding chloride equilibrium concentrations are listed in the last column in the above table.
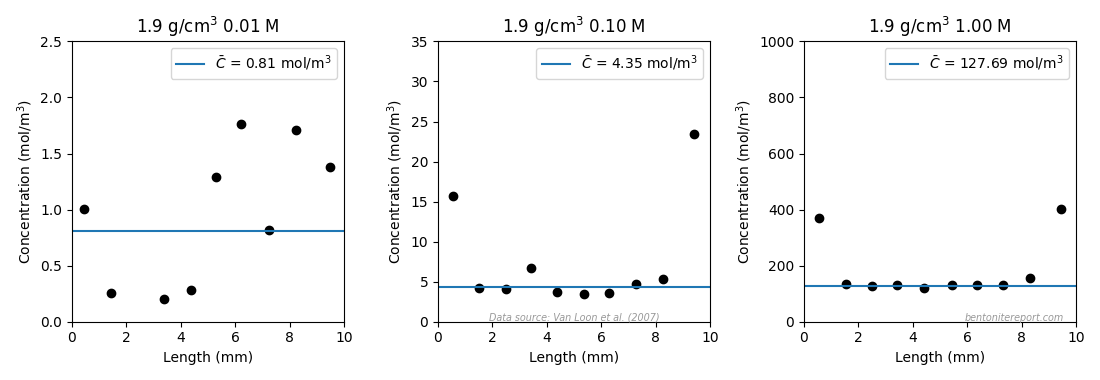
Finally, we also look at the 0.1 M test for which the steady-state tracer concentration profile was recorded. Extrapolating the linear part to the clay/source interface, gives a chloride content of 0.282 nmol/g, which corresponds to a clay concentration interval of 5.37⋅10-4 — 4.80⋅10-4 mol/m3, using the porosity interval defined above.9 Given the source concentration (0.024 mol/m3), these values correspond to a chloride equilibrium concentration ratio in the range 0.051 — 0.071.
The different ways of estimating chloride equilibrium concentrations provide a quite consistent picture (see above table). Although the information has been difficult to extract, it may thus seem that, in the end, all is good and well. However, we should note that the evaluated pore diffusivities show a quite peculiar dependency on background concentration.
Such a dependency, which has not been observed in earlier assessed studies, directly influence the evaluated equilibrium concentrations. As the breakthrough curves are so well sampled in the present study, this result can hardly be attributed to uncertainty in the values of \(D_p\). While Gl10 don’t explicitly identify this behavior (they do not evaluate \(D_p\)), a main focus of the study is actually to account for it, by means of “Archie’s law”, i.e. by suggesting a non-linear functional relationship between \(D_e\) and \(\epsilon_\mathrm{eff}\). I am strongly critical of such a treatment, but will refrain from discussing it here, as the focus of this assessment is the data itself rather than its interpretation (we have discussed this issue in a previous blog post).
An obvious alternative interpretation of this behavior is that chloride adsorbs on some system component, in the sense of becoming immobilized (what I have earlier dubbed true sorption). Gl11 test this hypothesis by performing additional batch sorption tests on the montmorillonite, in background solutions of NaCl and NaClO4 (0.5 M) at various pH. Although they cannot exclude a “\(R_d\)” value of the order of 10-4 m3/kg, they ultimately conclude that chloride do not sorb to any significant extent in these systems (and continues with “explaining” the behavior as resulting from other mechanisms).
I mean, however, that some experimental observations suggest that a sorption mechanism may be active. In addition to the above limit for the \(“R_d”\) value, we may note significant chloride sorption in the kaolinite samples, which were also studied in Gl10. There may of course be a reasonable explanation for why chloride sorption is observed in kaolinite, while it is not active in montmorillonite, but this issue is not really discussed in Gl10. Also, the recorded steady-state chloride content profile suggests a non-zero value at the interface to the target reservoir. This could, reasonably, indicate that some chloride is immobilized.
Perchlorate equilibrum concentrations
On the other hand, an additional argument against chloride sorption is that equilibrium perchlorate concentrations seem to be comparable with those evaluated for chloride. Gl11 don’t report perchlorate content directly, and we have to do some work to extract the corresponding equilibrium concentration in the 0.1 M sample that was sectioned. Gl11 plot the chloride tracer content for this sample together with “the concentration in the anion-accessible volume”, labelled \(c_\mathrm{acc}\).
\(c_\mathrm{acc}\) is, unsurprisingly, not a directly measured chloride concentration, but a quite elaborate interpretation of the data. From the unreported ClO4 content, an “anion-accessible porosity” variable has been calculated, by simply multiplying the physical porosity by the ratio between internal and external ClO4 concentrations. \(c_\mathrm{acc}\) is, in turn, defined as the actual measured chloride content distributed in a volume that corresponds to this “anion-accessible porosity”. By combining the reported chloride content (let’s call it \(\bar{n}_\mathrm{Cl}\)) and \(c_\mathrm{acc}\), we can thus de-derive the perchlorate equilibrium concentration as
Using this formula for the inner “linear” part of the profile (2 — 8 mm) gives the values 0.060, 0.059, 0.061 and 0.062, assuming nominal density. For porosity 0.394 the corresponding values are 0.044, 0.043, 0.044, and 0.045. We note that a range 0.043 — 0.062 for the equilibrium concentration ratio at 0.1 M background is in line with the previous evaluations. It should be noted, though, that this evaluation is for perchlorate, which not necessarily has the same equilibrium concentration as chloride. Nonetheless, this evaluation shows a similar, relatively high, equilibrium concentration also for this ion.
In fact, Gl11 provide results from yet another test where the focus is the perchlorate equilibrium,10 this time at a background concentration of 0.5 M. The results are reported as physical and “anion-accessible” porosities, evaluated from measuring water and perchlorate content.11
We note that also this sample shows substantial interface excess, but here we focus on the inner, relatively flat part (marked points in figure). From values of physical and effective porosity, we can directly calculate an equilibrium concentration in accordance with eq. 3. In this case the equilibrium concentration can also be related to a measured density. Using the average values gives a perchlorate equilibrium concentration ratio of \(\bar{c}_\mathrm{ClO_4}/0.5\; \mathrm{M} = 0.150\). Note that this value should be associated with density of 2.05 g/cm3 (the average porosity for the inner points is 0.259). This perchlorate equilibrium concentration ratio is nevertheless considerably larger than what was evaluated for chloride at (nominal) density 1.9 g/cm3 (0.11). This may indicate that perchlorate has a larger preference for the clay than chloride in these systems, but, as 2.05 g/cm3 is remarkably high, I suspect that measured water contents in this test have been systematically underestimated.
Summary and verdict
With only the information given in Gl10, I would judge the provided
information too uncertain to be used for quantitative process
understanding of chloride equilibrium in bentonite. With the
additional information provided in Gl11, however, we have seen that
the diffusion parameters — and consequently the equlibrium
concentrations that can be inferred — can be assessed to have been
quite robustly evaluated. Needless to say, access to a
completely separate publication should not be needed in order to make
this type of assessment. Nevertheless, my choice is to keep this data
to use for evaluating e.g. performance of models for salt exclusion.
A remaining uncertainty is the actual density of the tested samples. Results from corresponding water tracer tests suggest densities considerably lower than the nominal density. It not fully clear, however, if these water diffusion tests were conducted with separate samples or with the same samples as for the chloride diffusion tests.
Finally, these results complicate the picture of chloride equilibrium
concentrations in bentonite, as they do not fully comply with earlier
ones. In particular, here is observed a dependency of the pore
diffusivity on the background concentration, and chloride contents,
which are not seen in other studies. For anyone that is truly
interested in how salts distribute in bentonite, it should be a
priority to understand how the present results can be reconciled with
other chloride equilibirum results.12
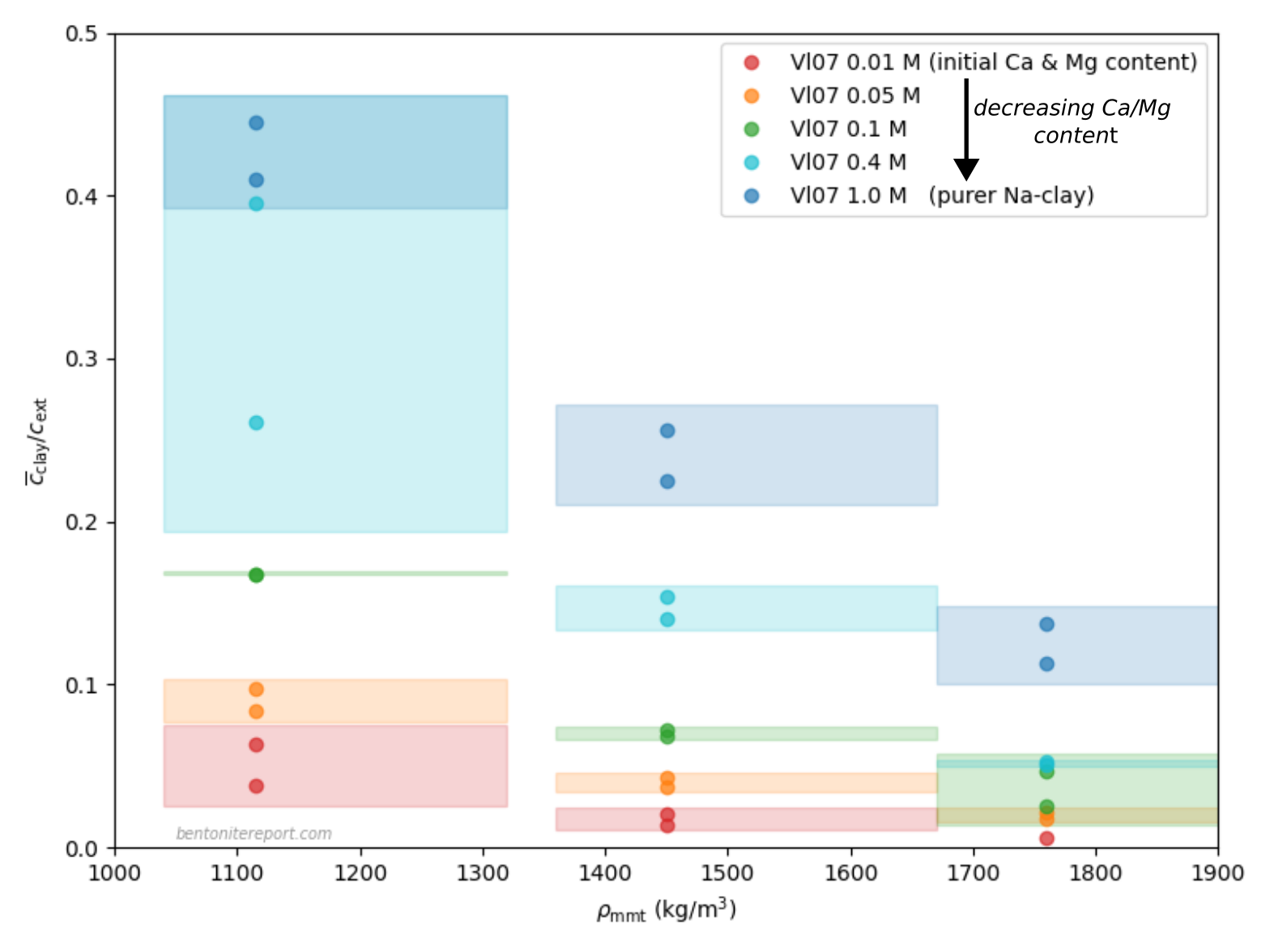
Below is plotted the chloride equilibrium concentrations evaluated
from this study. For each background concentration is drawn an
“uncertainty box”, that takes into account the uncertainty in
density, as discussed above, and the corresponding interval in
equlilibrium concentration ratio. The corresponding points have been
arbitrarily put in the middle of these “uncertainty boxes”. The
effective montmorillonite density has been calculated assuming a
montmorillonite content of 95%.
To compare the present results with others, we have also plotted some chloride equilibrium concentration evaluated from Van Loon et al. (2007), that we have assessed previously.
[1] To be fair, reading Gl10 carefully, out-diffusion is briefly mentioned a couple of times.
[2] Gl10 rather use the term “accessible porosity”, and symbol \(\epsilon_\mathrm{acc}\), but we stick with the terminology that we have used in thepreviousassessments. Also, a critique of mixing the effective porosity model (that involves \(\epsilon_\mathrm{eff}\)) and the traditional diffusion-sorption model (that involves \(\alpha\)) is found here.
[3] For background concentration 0.5 M it is difficult to resolve if the diagram in Gl10 has a single point, or if there are two points on top of each other. As Gl10 claim that duplicates were made at all concentrations, here we have assumed two different samples with identical parameters.
[4] The
through-diffusion flux evolution for background concentration 0.1 M
plotted in Gl10 seems not to be complete: the diagram shows data
points up until day 160, but Gl11 state that the test was conducted
for 229 days.
[5] The simulations presented here use \(L\) = 9.75 mm for the samples with background concentration 2.0 M, and \(L\) = 10.25 mm for the samples with background concentrations 0.1 M and 0.5 M. These are average values from the sample lenghts reported in Gl11.
Tracer profiles of 36Cl in Na–mom were found to be in qualitative agreement with those found by Molera et al. (2003) and exhibited two distinct linear regions with different slopes. In contrast to Molera et al. (2003) we interpret the 36Cl profiles in terms of heterogeneities of compaction in the boundary zones of the clays and not as the result of two diffusion processes. In view of these ambiguities, tracer profiles were generally used as a consistency test and not for the calculation of \(D_e\) values.
At least to me, this way of writing gives the impression that
profiles were recorded for most of the tests. In Gl11, however, we
learn that only a single profile was recorded.
[7] Gl11 argue for that the non-linear
parts of the profile actually reflect the state of the sample during
steady-state, rather than being an effect of dismantling. I am
strongly critical to their arguments, and plan to comment on this in
a separate blog post.
[8] For the sodium measurements in montmorillonite, it is certain that the above statement is false. Most of these were made in 5.4 mm samples, and they were all sectioned. Morover, these were reported in a much earlier publication: Glaus et al. (2007).
[9] The clay concentration is calculated as \(\bar{c} = \bar{n} \cdot \rho_d/\phi\), where \(\bar{n}\) denotes the chloride concentration as amount per dry mass.
[10] The main focus in Gl11 is
actually the density distribution in the interface regions of the
sample, but this is a straightforward perchlorate equilibrium test.
[11] The data in
this plot has been “de-scaled”, as it was measured in a 5.4 mm
sample, but then “recalculated” (!?) for a 10 mm sample in Gl11.
[12] I intend to write a
follow-up blog post discussing these issues.
The study consists of chloride and iodide though-diffusion tests in sets of samples of “Kunigel V1” bentonite, mixed with either 0%, 30%, or 50% silica sand. Here we mainly focus on the chloride tests. Also, we exclude the samples with 50% sand, as the montmorillonite content is judged to small. For each type of material, chloride diffusion tests were performed with NaNO3 background concentrations 0.01 M, 0.5 M, and 5.0 M. All samples are cylindrical with diameter 2 cm and height 1 cm (giving a volume of 3.14 cm3) and have dry density 1.6 g/cm3, which means that the effective montmorillonite density varies in the different test sets. To refer to a single test we use the notation “sand mixture percentage/background concentration”, e.g. “30/0.5” refers to the test made on the sample with 30% sand and with background concentration 0.5 M.
A single additional test was performed on purified “Kunipia F” material, at dry density 0.9 g/cm3 and a background concentration of 5.0 M NaNO3. This density was chosen in order to have a similar effective montmorillonite dry density as the “Kunigel V1” samples with 30% sand.
All tests were performed at elevated pH in the external solution of 12.5 (initially), and the Cl diffusion tests were performed in a N2 glove box, with vanishing CO2 and O2 pressures. In total we here investigate 7 tests (of the 22 tests in the full study, we exclude 12 that concern iodide diffusion, and 3 that have 50% sand). In addition to the published article, these tests are also reported in a technical report (in Japanese).
Materials
“Kunigel V1” and “Kunipia-F” are simply
brandnames rather than materials specifically aimed for scientific
studies. This is similar to e.g. “MX-80” and “KWK”, that we have
encountered in
previousassessments.
I have found it rather difficult to obtain official data on “Kunigel V1” and “Kunipia F”; data sheets or technical specifications do not seem readily available online. Moreover, the Japan Atomic Energy Agency seem to contain their data within a database, and restrict its usage (this site seems a bit deserted, though). Fortunately, the open scientific literature contains some entries. These sources, however, provide quite different values for e.g. montmorillonite content and exchangeable cations in “Kunigel V1”.
Montmorillonite content
Several studies of “Kunigel V1” — including Is08 — refer to a single source for e.g. mineral content and cation exchange capacity: Ito et al. (1994),1 which states that “Kunigel V1” contains 46% — 49% montmorillonite. Other sources, however, claim considerably different numbers; e.g. Cai et al. (2024) states a montmorillonite content of 54.3%, while Kikuchi and Tanai (2005) states 59.3%.
Here, I do not intend to critically assess these various sources, but simply conclude that the montmorillonite content stated in Is08 must be viewed with some skepticism. The study they reference (Ito et al. (1993)) is significantly older than their own, and they do not indicate that they have investigated the material actually used. In this assessment we adopt an uncertainty for the montmorillonite content in “Kunigel V1” of 45% — 60%.
Concerning “Kunipia F” most sources I have investigated state a montmorillonite content above 99%, although some — including Is08 — set a lower limit at 95%. Here we assume that the montmorillonite content of “Kunipia F” lies in the interval 95% — 100%.
Cation population
Reports on cation exchange capacity (CEC) and exchangeable cations in “Kunigel V1” are also quite scattered in the scientific literature, as demonstrated in the table below.
CEC values (roughly) in the range 0.55 — 0.80 eq/kg are reported. These numbers will not be further assessed here, and we will assume an uncertainty of this range for the CEC in “Kunigel V1”.
One observation to be made is that some of the sources reporting relatively high CEC also reports relatively high montmorillonite content. The data from e.g. Kikuchi and Tanai (2005) gives an estimate of the cation exchange capacity for the montmorillonite of 0.75/0.593 eq./kg = 1.26 eq./kg, while the data from Ito et al. (1994) gives roughly 0.556/0.475 eq./kg = 1.17 eq./kg. These numbers are quite consistent and suggest that the reported differences in CEC may partly be due to differences in montmorillonite content in different batches of “Kunigel V1”.
We can further conclude that the reported amount of exchangeable sodium in “Kunigel V1” is rather stable (with some exception), while the amount of exchangeable calcium and magnesium scatter significantly. This scatter is mainly due to interference of soluble accessory minerals (see below; entries in the above table where such interference is obvious are put within parentheses). Thus, the exchangeable cation population in “Kunigel V1” can be estimated to about 80% — 90% sodium and about 10% — 20% di-valent ions (calcium and magnesium).
Some cation data for “Kunipia F” found in the literature is listed in the table below (the table contains a few entries for the variants “Kunipia-G” and “Kunipia-P”; these are indicated).
The most commonly reported CEC value in this little survey is 1.19 eq./kg, and I suspect that this has been supplied by the manufacturer (although the value 1.15 eq./kg has also been reported as a given from the manufacturer). As “Kunipia F” is mainly pure montmorillonite, note that this value (1.19) is consistent with the montmorillonite CEC estimated from “Kunigel V1” above. That being said, the scatter in reported CEC for “Kunipia F” is in the range 1.0 — 1.22 eq./kg.
The few reported cation populations of “Kunipia F” (and the variant “Kunipia G”, which is supposed to be identical in composition) that I have found have a higher sodium content as compared with “Kunigel V1”, roughly in the range 85% — 95%.
Soluble accessory minerals
Basically all sources I have encountered — including Is08 — say that “Kunigel V1” contains smaller amounts of calcite and dolomite. This is also quite evident from some of the reported results on exchangeable cations, where the sum of these substantially exceeds the evaluated CEC. Obviously, the presence of additional calcium and magnesium contribute to the uncertainty and complexity when evaluating effects of ion equilibrium in this material (just as for the cases of “MX-80” and “KWK”).
Sample density
The samples in Is08 were ultimately sectioned and analyzed (for the final state concentration gradient). Is08 nowhere state that they measured density of these sections. We thus proceed with using the nominal density of 1.6 g/cm3. Using the above estimated uncertainty in montmorillonite content we get the following intervals for the effective montmorillonite density
Samples
EMMD interval (g/cm3)
0% sand
1.05 — 1.24
30% sand
0.83 — 1.01
Kunipia F
0.87 — 0.90
Note that these intervals do not include uncertainty due to variation in density of the actual samples.
Uncertainty of external solutions
The samples were prepared by first saturating them with deionized water for more than two weeks, and thereafter contacting them with NaNO3 solutions for more than five weeks.
We have no reason to doubt the accuracy of the initial concentration of the salt solutions, but contacting a bentonite containing di-valent ions with pure sodium solutions inevitably initiates an ion exchange process. We have made the same conclusion for studies using “MX-80” and “KWK” bentonite. Similar to the previous studies, Is08 do not keep track of the exact chemical evolution of the external solutions, but we can calculate an estimate of the extent of the sodium-for-di-valent exchange.
The above diagram shows the result of equilibrating the specified amount of bentonite (3.14 cm3) with the specified amount of external solution (100 ml) for different initial NaNO3 concentrations. The calculation assumes that the bentonite only contains sodium and calcium, with an initial calcium content of 15%, a selectivity coefficient of 5 M, and a cation exchange capacity of of 0.65 eq/kg. The diagram shows the amount of calcium left in the sample after equilibration, as a function of initial NaNO3 concentration for the cases of 0% and 30% mixed-in silica sand. The dashed vertical lines indicate the external concentrations in the performed tests. We note — as we have done for several other studies — that the equilibrium amount of di-valent ions still in the bentonite depends significantly on the initial NaNO3 concentration: tests performed at 0.5 M and 5.0 M gives essentially a pure sodium clay, while samples used at 0.01 M still contain the initial 15% di-valent ions in the clay.
Since the “Kunipia F” material only is used in a test with background concentration of 5.0 M, we can quite safely assume that the exchangeable cation population in this particular test is basically 100% sodium.
It should be noted that the calculations have not accounted for the
additional di-valent ions present in the bentonite in form of
accessory minerals (calcite, dolomite). They thus probably
underestimate the amount di-valent ions still left in the clay after
equilibration.
Evaluations from the diffusion tests
The diffusion tests were performed by sandwiching the clay samples between a source and target reservoir of equal volumes, 50 ml. The initial Cl tracer concentration was 0.05 mM in the source reservoir, and 0.0 mM in the target reservoir.
The tracer concentration in both the source and target reservoirs were
periodically measured, but as far as I understand, none of the
reservoir solutions were replaced during a test. This means that a
certain concentration build-up occurs in the target reservoir, and
a corresponding concentration drop occurs in the source reservoir.
The test set-up furthermore involves quite wide “filter” components at the interfaces between clay and reservoirs.2 Is08 mean that these components restrict diffusion to such an extent that they must be included in the test analyses. With a rather complex set-up that involves evolving reservoir concentrations and “filter” influence, the preferred way to evaluate them would be a full simulation of the whole process. This is however not the procedure followed in Is08 (below we make such simulations).
Instead, Is08 center most of their evaluation around the measured steady-state flux,3 taking filter diffusion into account. In the blog post on on filter influence on through-diffusion tests we derived an expression for the steady-state flux, which can be written
where \(D_e\) is the effective diffusivity, and \(L\) the length of the clay
component. \(\Delta c_\mathrm{res}\) is the difference in
concentration between the two reservoirs, and \(\omega\) is the relative
filter resistance, given by
\begin{equation} w = \frac{2D_eL_f}{D_fL} \tag{2} \end{equation}
where \(D_f\) and \(L_f\) denote effective diffusivity and length of the
two confining “filters” (assumed identical).
which is the same expression as found in Is08 (eqs. 2 and 3 in Is08).
\(D_e\) is thus evaluated in Is08 by measuring \(j^\mathrm{ss}\) and \(D_f\), estimating \(\Delta c_\mathrm{res}\), and knowing the lengths of the clay and filter components (\(L\) = 1 cm, \(L_f\) = 1.5 cm). Note that this is a quite involved procedure, necessitated by the test design: the source reservoir is small enough for the concentration to significantly drop during the course of a test; the target is not replaced during the course of a test, resulting in an increasing concentration significantly different from zero; the sample is sandwiched between wide “filter” components; and, as far as I can tell, the external solutions are not stirred or circulated. With a simpler test design, the reservoir concentration difference could have been kept effectively constant, and influence from confining filters could have been avoided (the only case, really, where filter influence is unavoidable is for cation through-diffusion at low ionic strength). With this being said, a re-evaluation of the results demonstrates that the “filter” influence, after all, is quite moderate. We will further discuss this below.
Is08 estimate \(\Delta c_\mathrm{res}\) by using the average source reservoir concentration during the course of a test (\(\bar{c}_\mathrm{source}\)), and by assuming zero target reservoir concentration, i.e. \(\Delta c_\mathrm{res} = 0-\bar{c}_\mathrm{source}\). I do not really understand this, because the target reservoir concentration is clearly not zero; since the two reservoirs have the same volume it seems more reasonable to assume that the concentration drop in the source reservoir corresponds to an equal concentration increase in the target reservoir.4
The “filter” diffusivities are claimed to be measured in separate tests without clay components, but the reported values does not make full sense to me. It is claimed that three different values for \(D_f\) were used for the three different background concentrations. But we do not expect any significant difference in diffusivity due to background concentration. Does this mean that tests performed at a specific background concetration all used the same test cell, while different test cells were used for different background concentrations? Furthermore, the specified values are \(D_f = 3\cdot 10^{-10}\) m2/s for background concentration 0.01 M, \(D_f = 2.6\cdot 10^{-9}\) m2/s for background concentration 0.5 M, and \(D_f = 1.8\cdot 10^{-9}\) m2/s for background concentration 5.0 M. The \(D_f\) values at high background concentration are thus not only almost an order of magnitude higher than that for 0.01 M background, these values also implies a diffusivity larger than for pure bulk water.5
If we anyway use these values for \(D_f\) to calculate the relative filter resistances (eq. 2) we get maximum values for \(\omega\) of 0.077, 0.037, and 0.055 for background concentrations 0.01 M, 0.5 M, and 5.0 M, respectively (anticipating the evaluated \(D_e\) values in table 1 in Is08). These values are tiny, showing that their own estimations indicate insignificant “filter” influence.
In the following we de-derive the values for \(j^\mathrm{ss}\) and \(\Delta c_\mathrm{b}\) (the final clay concentration difference) used for evaluating the reported values of \(D_e\), “\(D_a\)”, and \(\epsilon_\mathrm{eff}\),6 and compare them with the raw flux and concentration profile data (available for the tests performed with 30% sand mixture).
Steady-state fluxes
The steady-state fluxes are nowhere stated explicitly in Is08, but it is straightforward to read them off from the provided “breakthrough curves”. To check the consistency of the reported parameters we may use these values and the reported values for \(D_f\) and \(D_e\) to back-calculate \(\Delta c_\mathrm{res}\) using eq. 3.
In this table are also listed the “expected” values of of the reservoir concentration differences, \(\Delta c_\mathrm{res,ex}\), estimated from subtracting the average concentration increase in the target reservoir from from 0.05 mM. We see that the reported values of \(D_e\) “overestimates” \(\Delta c_\mathrm{res}\) by 8% — 40%.
We do not have more information to assess whether this mismatch is
due to some actual inconsistency in the reported values or if it indicates
that the concentration difference stated in the article was not
actually what was achieved in the experiment. In any case, this is low
quality scientific reporting.
Concentration profile gradients
We can, however, continue by also checking the consistency of the estimated pore diffusivity, \(D_p\),7 which was evaluated by measuring the concentration gradient in the clay at the termination of the tests (\(\Delta c_b/L\)).8
The concentration gradients are not explicitly stated in the article, but we can read them off from the published concentration plots. By using the tabulated values of \(D_p\) we can use eq. 4 to back-calculate what values for the steady-state flux was used for their evaluation.
Note that some of these values of \(j^\mathrm{ss}\) are smaller than
what can be read off from the “breakthrough curves”. In particular,
the value for the 30/5.0 test is reduced by more than 30%. If we use
these values of \(j^\mathrm{ss}\) to re-calculate the corresponding
reservoir concentration differences, we get
Although the calculated value for \(\Delta c_\mathrm{res}\) still is larger than 0.05 mM for the 30/0.5 test, these values are now generally in better agreement with the “expected” estimations.
I do not really know what to make of these results. For the 30/0.01 and 30/0.5 tests, the slightly different results perhaps reflect the uncertainty in the estimation of \(j^\mathrm{ss}\) and \(\Delta c_b\). But there is clearly something wrong with the evaluation of the 30/5.0 test. From the diagram (fig. 2 in Is08), it is, for example, clear that this test has the largest flux.
Chloride equilibrium concentrations
The chloride equilibrium concentration is evaluated in Is08 in terms of an “effective porosity,”6 \(\epsilon_\mathrm{eff} = D_e / D_p\). But from eq. 3 and eq. 4 we see that it is really evaluated from
Note that the factors \(j^\mathrm{ss}L\) cancel; the evaluation of \(\epsilon_\mathrm{eff}\) is therefore less sensitive to the estimation of \(j^\mathrm{ss}\) (the flux only appear in the correction term due to filter influence). Thus, even if the evaluation of \(j^\mathrm{ss}\) evidently has its flaws, the evaluation or \(\epsilon_\mathrm{eff}\) is more robust. This reflects the fact that the equilibrium concentration, as the name suggest, does not depend on transport quantities; as is clear from eq. 5, \(\epsilon_\mathrm{eff}\) is simply an interpretation of the clay concentration (\(c_b\)). We have discussed this issue severaltimesbefore.
Eq. 5 also shows that the uncertainty in estimating the equilibrium concentration (or \(\epsilon_\mathrm{eff}\)) mainly stem from uncertainties in \(\Delta c_\mathrm{res}\), and uncertainty stemming from filter resistance (\(2j^\mathrm{ss}L_f/D_f\)). Both of these uncertainties could have been avoided with a better test design — if filter resistance was avoided, and if the source and target reservoirs were kept at (virtually) constant concentrations, the equilibrium concentration would be given directly from the clay concentration profile.9
One way to estimate the effects of these uncertainties is to simply
compare the reported values for \(\epsilon_\mathrm{eff}\) with the ratio
\(\Delta c_b/\Delta c_\mathrm{res,init}\), where
\(\Delta c_\mathrm{res,init}\) = -0.05 mM is the initial reservoir
concentration difference.
The differences are not that great, demonstrating that reported values
of equilibrium concentrations (\(\epsilon_\mathrm{eff}\)) are quite
robust, even though we have found inconsistencies in the underlying
transport quantities.
Why not just simulate the whole thing?
A better way, in my view, to extract the equilibrium concentrations from this rather complex test set-up is to simulate the tests completely. This is done here, taking into account the external reservoir, the “filter” components and using the homogeneous mixture model for the bentonite component. Note that the homogeneous mixture and the effective porosity models are equivalent when it comes to modeling this type of diffusion: the effective porosity parameter can be calculated from \(\epsilon_\mathrm{eff} = \phi\cdot\Xi\), where \(\phi\) is the physical porosity and \(\Xi\) is the ion equilibrium coefficient. Similarly, the diffusion coefficient in the homogeneous mixture model (\(D_c\)) can in this case directly be identified with the pore diffusivity in the effective porosity model (\(D_p\)). In these simulations we used \(\Xi\) and \(D_c\) as fitting parameters.
The fitted parameters are listed in the table below and compared to
the reported values of \(D_p\) and \(\epsilon_\mathrm{eff}\).
Test
\(D_c\) (10-10 m2/s)
\(D_p\) (10-10 m2/s)
\(\Xi\)
\(\phi\cdot\Xi\)
\(\epsilon_\mathrm{eff}\)
30/0.01
2.14
1.8
0.103
0.043
0.043
30/0.5
2.50
2.4
0.457
0.19
0.13
30/5.0
2.39
1.6
0.625
0.26
0.21
Below is the simulated outflux curves and final state clay concentration profiles compared with experimental data.
0.01 M background concentration:
0.5 M background concentration:
5.0 M background concentration:
The simulations were performed both with (green lines) and without
(red lines) including filters. It may be noted that both models can be
fitted equally well, confirming that filter effects are after all
small in these tests. Also, although the diffusion parameters change
to some extent, the fitted ion equilibrium coefficients are
essentially the same regardless of whether filters are included or
not. We note that the spread in the values for the diffusion parameter
is smaller for the simulations as compared with the reported
values. As we expect similar diffusivity in these identically prepared
samples, I see this as a confirmation that a simulation better
captures the experimental parameters. Concerning the ion equilibrium,
or equivalently the “effective porosity”, we note that the
simulations provide somewhat higher values as compared with the
reported quantities, both for test 30/0.5 and test 30/5.0. The values
are however still comparable, again demonstrating that they have been
more robustly extracted.
Summary and verdict
We have seen that Is08 has several flaws and weaknesses: the test design is unnecessarily complex, and from the provided data on clay concentrations and fluxes, we have noted inconsistencies, e.g. in the values adopted for the steady state flux. It is also not completely clear if the actual initial concentration differences between the external reservoirs is 0.05 mM (as stated in the article) or if this is some measured but not reported quantity. We have also noted that the material used (primarily “Kunigel V1”) suffers from several uncertainties in its composition.
All of these factors lead to uncertainty in the quantities we are primarily interested in, i.e. chloride equilibrium concentrations. We have also seen, however, that we have reason to believe that these reported quantities are considerably more robust; most simplistically, the equilibrium concentration can be inferred by extrapolating the clay concentration profile to the interface on the target side and comparing that value to 0.05 mM.
My choice is therefore to keep these values to use for evaluating e.g. performance of models for salt exclusion. One reason that this data is interesting for this purpose is the measurement of equilibrium concentrations at an exceptionally high background concentration.
Below is a diagram that summarizes the findings of this assessment.
This figure includes gray stripes to indicate the estimated uncertainty in effective montmorillonite density (for these tests we have no means to estimate the uncertainty of the reported equilibrium concentration). For two of the tests that we have been able to look at in more detail (30/0.5 and 30/5.0) we have added an “area of uncertainty” that both include uncertainty in density and concentration. The estimation of the uncertainty in concentration is here simply done by including the values inferred from completely simulating these tests. These “areas” are no formal confidence intervals, but should be viewed as giving a hint of the uncertainties involved.
[1] Is08 actually refer to a corresponding technical report, from 1993.
[2] Apart from
“real” filters, the sample is also confined by two thick
perforated components; the total “filter” length is specified as
1.5 cm!
[3] As the concentration continually
changes in the reservoirs this is not a true steady-state, but what
we could call a “quasi”-steady state (it is still easily
distinguished from the initial part of a through diffusion test).
[4] With reservation for that the target is consumed
due to quite frequent sampling — but this would contribute to an
additional increase of the target concentration.
[5] Note that these are effective diffusivities, and includes “filter” porosity (we are not told their values, but they can of course not be larger than unity). The only value of \(D_f\) that seems reasonable is the one for 0.01 M, which corresponds to a geometric factor of 6.7. To make things even stranger, for iodide the same value is used for \(D_f\) regardless of background concentration (\(3.9\cdot 10^{-10}\) m2/s).
[6] Is08 refer to this quantity as \(\alpha\), “the capacity factor”. But it is clear from the text that it is interpreted as an effective porosity, and we will therefore use the notation \(\epsilon_\mathrm{eff}\), in accordance with earlier assessments. Is08 actually also relate the parameter \(\alpha\) to sorption via the relation \(\alpha = \phi + \rho K_d\) (their eq. 6). This is however a mix-up of two incompatible models, which I have commented on here. We also note that Is08 actually never use the distribution coefficient, \(K_d\), for anything in their analysis.
[7] Is08 call this quantity \(D_a\), but it is not an “apparent” diffusivity, and I do not accept using this bizarre nomenclature. I will call the corresponding parameter \(D_p\) in accordance with e.g. the effective porosity model.
[8] Is08 define the clay concentration, \(c_b\), in terms of total clay volume. Alternatively it can be defined in terms of total amount of water, the difference being a porosity factor.
[9] Or, rather, chloride equilibrium concentrations can then be inferred directly from the value of the clay concentration profile at the interface to the source reservoir.
Here’s an opinion: The compacted bentonite research field is currently
in a terrible state.
After a period away, I’ve recently begun catching up on newly published research in this field. With a fresh perspective, yet still influenced by writing over 30 long-reads over the past years, I can’t help but wonder: what is the problem? Why are a majority of researchers stuck with a view of bentonite1 that essentially makes no sense? And why has this view been the mainstream for decades now?
I get how this might come across: a solitary man ranting on a blog,
criticizing an entire research field in less-than-perfect English. I
probably smell bad and have some wild ideas about why General
Relativity is wrong as well. But what I’m aiming for with this blog
is simply a platform to present an alternative to the mainstream,
primarily because it annoys me as a science-minded person how absurd
this view is.2 I
understand that I will likely struggle to convince anyone who is
already invested in this view, but I’m trying to put myself in the
shoes of e.g. someone entering this field for the first time.
For these reasons, I will try something a little new here: reviewing already published papers. I have touched on this in various forms before, but then usually with a broader topic in mind. Now I intend to critically assess specific publications from the outset. As a first publication to review in this way, I have chosen “Ionic Transport in Nano-Porous Clays with Consideration of Electrostatic Effects” (Tournassat and Steefel, 2015), for the following reasons
It is published in “Reviews in Mineralogy & Geochemistry”, which claims that “The content of each volume consists of fully developed text which can be used for self-study, research, or as a text-book for graduate-level courses.” If anyone aims to learn about ion transport in bentonite from this publication, I would certainly recommend to also consider this review.
It is a quite comprehensive source for many of the claims of the contemporary mainstream view that I have described in earlier blog posts. I guess it makes sense for a publication in “Reviews in Mineralogy & Geochemistry” to reflect the typical view of a research field.
It is published as open access. The article is thus accessible to anyone who wants to check the details.
I will use the abbreviation TS15 in following to refer to this
publication.
Overview
The article covers 38 journal pages (+ references) and includes quite
a lot of topics. At the highest level of headings, the outline look
like this
Introduction (p. 1 — 2)
Classical Fickian Diffusion Theory (p. 2 — 9)
Clay mineral surfaces and related properties (p. 9 — 17)
Constitutive equations for diffusion in bulk, diffuse layer, and interlayer water (p. 17 — 23)
Relative contributions of concentration, activity coefficient and diffusion potential gradients to total flux (p. 24 — 28)
From diffusive flux to diffusive transport equations (p. 28 — 33)
Applications (p. 33 — 37)
Summary and Perspectives (p. 37 — 38)
Given the quite large scope of TS15, I will present this review in
parts, with this first part focusing on the introduction and the
section titled “Classical Fickian Diffusion Theory”.
“Introduction”
I find it remarkable that the authors use terms like “clays” and “clay minerals” when speaking of properties such as “low permeability”, “high adsorption capacity” and “swelling behavior”, and of applications such as nuclear waste storage. I mean that using such general terms here is too broad, as the article focuses solely on systems with swelling/sealing ability. Such an ability is generally connected with a significant cation exchange capacity. Here, I will refer to such systems as “bentonite”, although I am aware that I use the term quite sloppily. But I think this is better than to refer to the components as general “clay minerals” — I don’t think anyone consider it a good idea to e.g. use talc or kaolinite as buffer materials in nuclear waste repositories. Moreover, most of the examples considered in the article are systems that can be described as bentonite. Given the title of the article I also expect a definition of “nano-porous clays”. It is not given here, and the term is actually not used at all in the entire text! (Except one time at the very end.)
After providing a brief overview of the application of (sealing) clay materials, the introduction takes, in my opinion, a rather drastic turn (it happens without even changing paragraphs!).
Clay transport properties are however not simple to model, as they deviate in many cases from predictions made with models developed previously for “conventional” porous media such as permeable aquifers (e.g., sandstone). […] In this respect, a significant advantage of modern reactive transport models is their ability to handle complex geometries and chemistry, heterogeneities and transient conditions (Steefel et al. 2014). Indeed, numerical calculations have become one of the principal means by which the gaps between current process knowledge and defensible predictions in the environmental sciences can be bridged (Miller et al. 2010).
I think the first sentence is too subjective and general. Given the above discussion, here the term “clay transport properties” can cover a million things, if read at face value. Are all of them difficult to model? Also, something does not have to be more difficult just because it deviates from the “convention”. I would argue that several aspects of bentonite actually make it easier to model than, say, sandstone. Advective processes, for example, can often be neglected in compacted bentonite.
I find the statement regarding the advantage of reactive transport
models highly problematic. Not only does it read more like an
advertisement for the authors’ own tools than “fully developed text
for self-study”, but the authors also seem ignorant of issues like
the dangers of overparameterization (a theme that will recur).
“Classical Fickian Diffusion Theory”
As the title of the next section is “Classical Fickian Diffusion Theory,” a reader expects a discussion focused solely on diffusive process, especially when the immediate subtitle reads “Diffusion Basics.” I therefore find it peculiar that this section actually presents the traditional diffusion-sorption model, which describes a combination of diffusion and sorption processes. The model is summarized in eq. 10 in TS15
where \(c\) is the “pore water” concentration of the considered species, \(D_e\) its “effective diffusivity”, \(K_D\) the sorption partition coefficient, \(\rho_d\) dry density, and \(\phi\) porosity.3 For later considerations we also note that TS15 define the denominator on the right hand side as the “rock capacity factor”, \(\alpha = \phi + \rho_dK_D\).
I find it particularly odd that two of the fundamental assumptions of
this specific model are essentially left uncommented, namely that
sorbed ions are immobilized and that the pores contain bulk
water. Instead, the authors appear to question the assumption of
Fickian diffusion in the context of clay systems, i.e. that diffusive
fluxes are assumed proportional to corresponding aqueous concentration
gradients.
This section aims, as far as I can see, to point out shortcomings in the description of diffusion in bentonite, and to motivate further model development. But it should be clear from the outset that using the traditional diffusion-sorption model as the basis for such an endeavor is doomed to fail. The reason for this failure is not due to assuming Fickian diffusion, but due to the other two model assumptions; it has long been demonstrated that exchangeable ions are mobile, and the notion that compacted bentonite contains mainly bulk water is absurd.
After the traditional diffusion-sorption model has been presented, it is evaluated by investigating how it can be fitted to tracer through-diffusion data (this is restatement of original work of Tachi and Yotsjui (2014)). Not surprisingly, it turns out that fitted diffusion coefficients may be unrealistically large. This is of course a direct consequence of the incorrect assumption of immobility in the traditional diffusion-sorption model. TS15 also appear to dismiss the model, saying
This result […] is not physically correct and points out the
inconsistency of the classic Fickian diffusion theory for modeling
diffusion processes in clay media.
I am bothered, though, that they keep using the phrase “classic Fickian diffusion theory”, which inevitably focuses on the Fickian aspect rather than on the obviously incorrect assumptions of the chosen model. Also, rather than simply concluding that the model is incorrect, TS15 continues4
[T]he large changes of \(\mathrm{Cs}^+\) diffusion parameters as a function of chemical conditions (\(D_{e,\mathrm{Cs}^+}\) decreases when the ionic strength increases […]) highlight the need to couple the chemical reactivity of clay materials to their transport properties in order to build reliable and predictive diffusion models.
There is no rationale for such a conclusion. I don’t even completely
understand what “couple the chemical reactivity of clay materials to
their transport properties” mean. Isn’t that what the traditional
diffusion-sorption model attempts? What unrealistic \(D_e\) values
actually highlights is simply that one should not use a model that
assumes immobilization of “sorbed” ions.
However, the experimental observations were completely different: \(^{22}\mathrm{Na}^+\) accumulated in the high NaCl concentration reservoir as it was depleted in the low NaCl concentration reservoir, evidencing non-Fickian diffusion processes.
This is plain wrong. As explained in detail in an earlier post, the diffusion process in the “uphill” test is certainly Fickian. What the test demonstrates is, again, that “sorbed” ions are not immobile.
TS15 also comment on the results of fitting the model to anion tracer through-diffusion data. Here, as is well known, the fitted “rock capacity factor” \(\alpha = \phi + \rho_dK_D\) becomes significantly lower than the porosity \(\phi\). From the perspective of the traditional diffusion-sorption model, this is completely infeasible, as it implies a negative \(K_D\). But rather than simply dismissing the model, TS15 state
The lower \(\alpha\) values for anions than for water indicate that
anions do not have access to all of the porosity.
Also this is incorrect. The porosity5 is an input
parameter rather than a fitting parameter in the traditional
diffusion-sorption model. When claiming that a small value of \(\alpha\)
indicates a decreased porosity, TS15 reinterpret the parameter,
on the fly, in terms of a completely different model:
the effective porosity model. This model has not been mentioned at
all earlier in the article.6
As has been discussed earlier on the blog, the effective porosity model can be fitted to anion tracer through-diffusion data, but now we need to keep track of two different models in the evaluation (something that TS15 do not). Moreover, these two models (the traditional diffusion-sorption and the effective porosity models) are incompatible. But TS15 continue by saying
This result is a first direct evidence of the limitation of the
classic Fickian diffusion theory when applied to clay porous media:
it is not possible to model the diffusion of water and anions with
the same single porosity model. The observation of a lower \(\alpha\)
value for anions than for water led to the development of the
important concept of anion accessible porosity […]
This is a terrible passage. To begin with, the “Fickian” aspect is
also here implied as the problem. But the reason for why the
traditional diffusion-sorption model cannot be fitted to anion tracer
through-diffusion data is of course because this model assumes the
entire pore space to be filled with bulk water. Further, it’s hardly
comprehensible what the authors mean by “it is not possible to model
the diffusion of water and anions with the same single porosity
model”. I think they simply mean that for water you must choose
\(\alpha = \phi\), while for anion through-diffusion you instead must
“choose” \(\alpha < \phi\). But the result \(\alpha < \phi\) should only
lead to the conclusion that the traditional diffusion-sorption model
cannot in any reasonable sense be fitted. A favorable reading of this
passage is to assume that the authors actually mean that the
effective porosity model can only be fitted to anion and water
tracer through-diffusion data by using different values of the
(effective) porosity, and that any “rock capacity factor” should not
appear in this discussion.
Finally, the last sentence gives me headache. Rather than being an “important concept”, I mean that the idea of an “anion accessible porosity” has caused tremendous damage to the development of the bentonite research field for several decades now. We have earlier discussed on the blog that the whole idea of “anion accessible porosity” is based on misunderstandings. We have also demonstrated that the effective porosity model is not valid, even though it can be fitted to anion tracer through-diffusion data. A simple way to see this is to consider closed-cell diffusion data rather than through-diffusion data. Closed-cell tests are simpler than through-diffusion tests, as they don’t involve interfaces between clay and external solutions. We can e.g. take a look at the vast amount of diffusion coefficients for chloride in montmorillonite, presented in Kozaki et al. (1998).
There are in total 55(!) values, corresponding to 55(!) separate tests. These have been systematically varied with respect to density and temperature, but all of them were performed on montmorillonite equilibrated with distilled water. From the perspective of the effective porosity model, the effective porosity in such a system should be minute, perhaps even strictly zero; effective porosities evaluated from chloride through-diffusion tests are well below 1% even at a background concentration as large as 10 mM. Thus, if the idea of “anion accessible porosity” was reasonable, we’d expect extremely low values of the chloride diffusion coefficient in the above plot.7 We’d perhaps also expect a threshold behavior, where chloride diffusivity basically vanishes above a certain density. But this is not at all the behavior: chloride is seen to diffuse just fine in all 55(!) tests, with temperature- and density dependencies that seems reasonable for a homogeneous system. Moreover, chloride behaves very similarly to e.g. sodium, as seen here
Here the sodium data is from Kozaki et al. (1998),8 and it has also been measured in montmorillonite equilibrated with distilled water.
The effective porosity model and the notion of “anion accessible porosity” can consequently be dismissed directly, by comparing with simpler tests than what is done in TS15. The reason that the effective porosity model can be fitted to anion through-diffusion data must be attributed to a misinterpretation of such tests, as they involve also interfaces to external solutions. At least to me it is completely clear that what many researchers interpret as an effective porosity is actually effects of interface equilibrium.
If TS15 were serious about evaluating bentonite diffusion processes in
this section I think they should have done the following:
Discuss the assumptions of ion immobility of sorbed ions and bulk pore water when presenting the traditional diffusion-sorption model. Moreover, they should not call this “Classical Fickian Diffusion Theory”.
Also present and discuss the effective porosity model, as they obviously use it in their evaluations. They actually even seem to promote it! And it is as “Fickian” as the traditional diffusion-sorption model.
Evaluate the models using closed-cell data to avoid misinterpretations arising from complications at bentonite/external solution interfaces.
Conclude that the traditional diffusion-sorption model is not valid for bentonite, and that this is because of the assumptions of immobility of sorbed ions and bulk pore water.
Conclude that the effective porosity model is not valid for bentonite, and that the notion of “anion accessible porosity” is flawed.
Instead, we get a quite confused and incomplete description, mixed
with entirely inaccurate statements. In the end, it is difficult to
understand what the takeaway message of this section really is. A
reader is left with an impression that there is some problem with the
“Fickian” aspect of diffusion, but nothing is spelled out. We have
also been hinted that “anion accessible porosity” is important,
without really having been introduced to the concept/model.
The section ends with the following passage
The limitations of the classic Fickian diffusion theory must find
their origin in the fundamental properties of the clay minerals. In
the next section, these fundamental properties are linked
qualitatively to some of the observations described above.
If “classic Fickian diffusion theory” here is interpreted as “the traditional diffusion-sorption model” (which is literally what has been presented), the first sentence is both incorrect and trivial at the same time. The traditional diffusion-sorption model does not have “limitations” — it is fundamentally incorrect as a model for bentonite. The reason for this is that exchangeable ions are not immobile and that bentonite does not contain significant amounts of bulk water. Both of these reasons can be linked to “fundamental properties” of some specific clay minerals.
But it is clear that TS15 also have vaguely promoted the concept of “anion accessible porosity” and the effective porosity model. Are these not included in “the classic Fickian diffusion theory”? If not, why then is a model that assumes sorption and immobilization?
How can it not be immediately obvious to everyone that the
diffusion process is much simpler than the contemporary descriptions?
As we have brought up the data from Kozaki etal. (1998), I would like to end this blog post by further considering actual profiles of chloride and sodium diffusing in montmorillonite.
This figure shows the corresponding normalized concentration profiles after 23.7 hours in closed-cell tests performed at \(50\;^\circ\mathrm{C}\) in Na-montmorillonite at dry density \(1.8 \;\mathrm{g/cm^3}\) that has been equilibrated with distilled water. In the case of sodium, both the profile evaluated from Fick’s second law (orange line) and measured values (circles) are plotted. In the case of chloride, no measured values are available, but the value of the diffusion coefficient is the result of fitting Fick’s second law (green line) to such data.
From the perspective of the traditional diffusion-sorption model, the
sodium profile is supposed to represent the combined result of ions
diffusing in bulk water, at a rate many orders of magnitude larger
than in pure water, while being strongly retarded due to sorption onto
“the solid” (where the ions are immobile). This is clearly nonsense,
and something that I think TS15 actually tries to communicate.
From the perspective of the effective porosity model, on the other
hand, the chloride profile is supposed to be the result of the ion
diffusing in an essentially infinitesimal fraction of the pore volume,
which magically is perfectly interconnected in all samples on which such
tests are conducted. This is of course just as nonsensical as
the above interpretation of the sodium profile, but in this case TS15
appear to promote the model (the “important concept of anion
accessible porosity”).
Note that these two simple ions, at the end of the day, diffuse very similarly (please stop reading for a moment and contemplate the above plot). If sodium and chloride actually migrate in completely different domains and are subject to completely different physico-chemical processes, this “coincident” would be more than a little weird. Especially given that the two ions show similar diffusive behavior across a wide range of densities. To me, this simple observation makes it evident that ion diffusion in bentonite at the basic level is much simpler than what is suggested by the contemporary mainstream view. I mean that it is completely obvious that all ions in bentonite diffuse in the same type of quite homogeneous domain. And since it cannot be argued that the pore volume is dominated by anything other than interlayers at 1.8 g/cm³, this homogeneous domain is the interlayer domain at any relevant density. The evidence has been available for at least 25 years (in fact much longer than that). How can this be difficult to grasp?
Update (250213): Part II of this review is found here.
Footnotes
[1] By “bentonite” I here mean any type of smectite-rich system with a significant cation exchange capacity.
[2] The irony is that the “alternative” in a
broader perspective is more mainstream than the “mainstream”
view. I basically propose to obey the laws of thermodynamics.
[3] I have simplified the notation here somewhat compared with how it is written in TS15. As many others, TS15 call this equation “Fick’s second law” (via their eq. 4), which is not correct. Fick’s laws refer strictly to pure diffusion processes. However, the equation has the same form as Fick’s second law, if \(D_e/(\phi + \rho_d K_D)\) is treated as a single constant (often referred to as the apparent diffusivity).
[4] This behavior is of course not
unique for cesium; I don’t know why TS15 focus so hard on that ion
here.
[5] “Porosity” is a volume ratio. I’m not a fan of that the word has also begun to mean “pore space” in the bentonite scientific literature.
[6]
In fact, \(\alpha\) has earlier in the article been unambiguously
related to sorption:
If the species \(i\) is also adsorbed on or
incorporated into the solid phase, then it is possible to define a
rock capacity factor \(\alpha_i\) that relates the concentration in
the porous media to the concentration in solution
[7] That the diffusivity is much too large for an effective porosity interpretation to make sense can also be seen from invoking Archie’s law, which is quite popular in bentonite scientific papers.
Here \(D_0\) is the diffusivity in pure bulk water, which is about \(2\cdot 10^{-9} \;\mathrm{m^2/s}\) for chloride. Using the popular choice \(n \approx 2\) and choosing e.g. \(\epsilon_\mathrm{eff} = 0.001\) (most probably an overestimation when using distilled water), we get
This is more than twenty times the actual value for \(D_0\). (\(D_e = 5.1\cdot 10^{-14} \;\mathrm{m^2/s}\) is evaluated from Kozaki’s data at \(1.4 \;\mathrm{g/cm^3}\) and \(25\;^\circ\mathrm{C}\))
[8] Note! This publication is different from the
chloride study.
Vl07 is centered around a set of through-diffusion tests in “KWK” bentonite samples of nominal dry densities 1.3 g/cm3, 1.6 g/cm3, and 1.9 g/cm3. For each density, chloride tracer diffusion tests were conducted with NaCl background concentrations 0.01 M, 0.05 M, 0.1 M, 0.4 M, and 1.0 M. In total, 15 samples were tested. The samples are cylindrical with diameter 2.54 cm and height 1 cm, giving an approximate volume of 5 cm3. We refer to a specific test or sample using the nomenclature “nominal density/external concentration”, e.g. the sample of density 1.6 g/cm3 contacted with 0.1 M is labeled “1.6/0.1”.
After maintaining steady-state, the external solutions were replaced
with tracer-free solutions (with the same background concentration),
and tracers in the samples were allowed to diffuse out. In this way,
the total tracer amount in the samples at steady-state was
estimated. For tests with background concentrations 0.01 M, 0.1 M, and
1.0 M, the outflux was monitored in some detail, giving more
information on the diffusion process. After finalizing the tests, the
samples were sectioned and analyzed for stable (non-tracer)
chloride. In summary, the tests were performed in the following
sequence
Saturation stage
Through-diffusion stage
Transient phase
Steady-state phase
Out-diffusion stage
Sectioning
Uncertainty of samples
The used bentonite material is referred to as “Volclay KWK”. Similar to “MX-80”, “KWK” is just a brand name (it seems to be used mainly in wine and juice production). In contrast to “MX-80”, “KWK” has been used in only a fewresearchstudies related to radioactive waste storage. Of the studies I’m aware, only Vejsada et al. (2006) provide some information relevant here.1
Vl07 state that “KWK” is similar to “MX-80” and present a table with chemical composition and exchangeable cation population of the bulk material. As the chemical composition in this table is identical to what is found in various “technical data sheets”, we conclude that it does not refer to independent measurements on the actual material used (but no references are provided). I have not been able to track down an exact origin of the stated exchangeable cation population, but the article gives no indication that these are original measurements (and gives no reference). I have found a specification of “Volclay bentonite” in this report from 1978(!) that states similar numbers (this document also confirms that “MX-80” and “KWK” are supposed to be the same type of material, the main difference being grain size distribution). We assume that exchangeable cations have not been determined explicitly for the material used in Vl07.
In a second table, Vl07 present a mineral composition of “KWK”, which I assume has been determined as part of the study. But this is not fully clear, as the only comment in the text is that the composition was “determined by XRD-analysis”. The impression I get from the short material description in Vl07 is that they rely on that the material is basically the same as “MX-80” (whatever that is).
Montmorillonite content
Vl07 state a smectite content of about 70%. Vejsada et al. (2006), on the other hand, state a smectite content of 90%, which is also stated in the 1978 specification of “Volclay bentonite”. Note that 70% is lower and 90% is higher than any reported montmorillonite content in “MX-80”. Regardless whether or not Vl07 themselves determined the mineral content, I’d say that the lack of information here must be considered when estimating an uncertainty on the amount of montmorillonite (“smectite”) in the used material. If we also consider the claim that “KWK” is similar to “MX-80”, which has a documented montmorillonite content in the range 75 — 85%, an uncertainty range for “KWK” of 70 — 90% is perhaps “reasonable”.
Cation population
Vl07 state that the amount exchangeable sodium is in the range 0.60 — 0.65 eq/kg, calcium is in the range 0.1 — 0.3 eq/kg, and magnesium is in the range 0.05 — 0.2 eq/kg. They also state a cation exchange capacity in the range 0.76 — 1.2 eq/kg, which seems to have been obtained from just summing the lower and upper limits, respectively, for each individual cation. If the material is supposed to be similar to “MX-80”, however, it should have a cation exchange capacity in the lower regions of this range. Also, Vejsada et al. (2006) state a cation exchange capacity of 0.81 eq/kg. We therefore assume a cation exchange capacity in the range 0.76 — 0.81, with at least 20% exchangeable divalent ions.
Soluble accessory minerals
According to Vl07, “KWK” contains substantial amounts of accessory carbonate minerals (mainly calcite), and Vejsada et al. (2006) also state that the material contains calcite. The large spread in calcium and magnesium content reported for exchangeable cations can furthermore be interpreted as an artifact due to dissolving calcium- and magnesium minerals during the measurement of exchangeable cations (but we have no information on this measurement). Vl07 and Vejsada et al. (2006) do not state any presence of gypsum, which otherwise is well documented in “MX-80”. I do not take this as evidence for “KWK” being gypsum free, but rather as an indication of the uncertainty of the composition (the 1978 specification mentions gypsum).
Sample density
Vl07 don’t report measured sample densities (the samples are ultimately sectioned into small pieces), but estimate density from the water uptake in the saturation stage. The reported average porosity intervals are 0.504 — 0.544 for the 1.3 g/cm3 samples, 0.380 — 0.426 for the 1.6 g/cm3 samples, and 0.281 — 0.321 for the 1.9 g/cm3 samples. Combining these values with the estimated interval for montmorillonite content, we can derive an interval for the effective montmorillonite dry density by combining extreme values. The result is (assuming grain density 2.8 g/cm3, adopted in Vl07).
Sample density (g/cm3)
EMDD interval (g/cm3)
1.3
1.04 — 1.32
1.6
1.36 — 1.67
1.9
1.67 — 1.95
These intervals must not be taken as quantitative estimates, but as giving an idea of the uncertainty.
Uncertainty of external solutions
Samples were water saturated by first contacting them from one side with the appropriate background solution (NaCl). From the picture in the article, we assume that this solution volume is 200 ml. After about one month, the samples were contacted with a second NaCl solution of the same concentration, and the saturation stage was continued for another month. The volume of this second solution is harder to guess: the figure shows a smaller container, while the text in the figure says “200 ml”. The figure shows the set-up during the through-diffusion stage, and it may be that the containers used in the saturation stage not at all correspond to this picture. Anyway, to make some sort of analysis we will assume the two cases that samples were contacted with solutions of either volume 200 ml, or 400 ml (200 ml + 200 ml) during saturation.
The through-diffusion tests were started by replacing the two saturating solutions: on the left side (the source) was placed a new 200 ml NaCl solution, this time spiked with an appropriate amount of 36Cl tracers, and on the right side (the target) was placed a fresh, tracer free NaCl solution of volume 20 ml. The through-diffusion tests appear to have been conducted for about 55 days. During this time, the target solution was frequently replaced in order to keep it at a low tracer concentration. The source solution was not replaced during the through-diffusion test.
As (initially) pure NaCl solutions are contacted with bentonite that contains significant amounts of calcium and magnesium, ion exchange processes are inevitably initiated. Thus, in similarity with some of the earlierassessed studies, we don’t have full information on the cation population during the diffusion stages. As before, we can simulate the process to get an idea of this ion population. In the simulation we assume a bentonite containing only sodium and calcium, with an initial equivalent fraction of calcium of 0.25 (i.e. sodium fraction 0.75). We assume sample volume 5 cm3, cation exchange capacity 0.785 eq/kg, and Ca/Na selectivity coefficient 5.
Below is shown the result of equilibrating an external
solution of either 200 or 400 ml with a sample of density 1.6 cm3/g,
and the corresponding result for density 1.3 cm3/g and external volume
400 ml. As a final case is also displayed the result of first
equilibrating the sample with a 400 ml solution, and then replacing it
with a fresh 200 ml solution (as is the procedure when the
through-diffusion test is started).
Although the results show some spread, these simulations make it relatively clear that the ion population in tests with the lowest background concentration (0.01 M) probably has not changed much from the initial state. In tests with the highest background concentration (1.0 M), on the other hand, significant exchange is expected, and the material is consequently transformed to a more pure sodium bentonite. In fact, the simulations suggest that the mono/divalent cation ratio is significantly different in all tests with different background concentrations.
Note that the simulations do not consider possible dissolution of accessory minerals and therefore may underestimate the amount divalent ions still left in the samples. We saw, for example, that the material used in Muurinen et al. (2004) still contained some calcium and magnesium although efforts were made to convert it to pure sodium form. Note also that the present analysis implies that the mono/divalent cation ratio probably varies somewhat in each individual sample during the course of the diffusion tests.
Direct measurement of clay concentrations
Chloride
clay concentration profiles were measured in all samples after
finishing the diffusion tests, by dispersing sample sections in
deionized water. Unfortunately, Vl07 only present this chloride
inventory in terms of “effective” or
“Cl-accessible porosity”, a concept often encountered in
evaluation of diffusivity. However, “effective porosity” is
not what is measured, but is rather an interpretation of
the evaluated amount of chloride in terms of a certain pore volume
fraction. Vl07 explicitly define effective porosity as
\(V_\mathrm{Cl}/V_\mathrm{1g}\), where \(V_\mathrm{1g}\) is the “volume
of a unit mass of wet bentonite”, and \(V_\mathrm{Cl}\) is the “volume
of the Cl-accessible pores of a unit mass of bentonite”. While
\(V_\mathrm{1g}\) is accessible experimentally, \(V_\mathrm{Cl}\) is
not. Vl07 further “derive” a formula for the effective porosity
(called \(\epsilon_\mathrm{eff}\) hereafter)
where \(n’_\mathrm{Cl}\) is the amount chloride per mass bentonite, \(\rho_\mathrm{Rf}\) is the density of the “wet” bentonite, and \(C_\mathrm{bkg}\) is the background NaCl concentration.2 In contrast to \(V_\mathrm{Cl},\) these three quantities are all accessible experimentally, and the concentration \(n’_\mathrm{Cl}\) is what has actually been measured. For a result independent of how chloride is assumed distributed within the bentonite, we thus multiply the reported values of \(\epsilon_\mathrm{eff}\) by \(C_\mathrm{bkg}\), which basically gives the (experimentally accessible) clay concentration
Here we also have divided by sample porosity, \(\phi\), to relate the clay concentration to water volume rather than total sample volume. Note that eq. 2 is not derived from more fundamental quantities, but allows for “de-deriving” a quantity more directly related to measurements. (I.e., what is reported as an accessible volume is actually a measure of the clay concentration.)
It is, however, impossible (as far as I see) to back-calculate the actual value of \(n’_ \mathrm{Cl}\) from provided formulas and values of \(\epsilon_\mathrm{eff}\), because masses and volumes of the sample sections are not provided. Therefore, we cannot independently assess the procedure used to evaluate \(\epsilon_\mathrm{eff}\), and simply have to assume that it is adequate.3 Here are the reported values of \(\epsilon_\mathrm{eff}\) for each test, and the corresponding evaluation of \(\bar{C}\) using eq. 2 (column 3)
*) The table in Vl07 says 0.076, but the concentration profile diagram says 0.090. **) The table in Vl07 says 0.16, but this must be a typo.
When using eq. 2 we have adopted porosities 0.536, 0.429, and 0.322,
respectively, for densities 1.3 g/cm3, 1.6 g/cm3, and 1.9 g/cm3.
The tabulated \(\epsilon_\mathrm{eff}\) values are evaluated as averages of the clay concentration profiles (presented as effective porosity profiles), which look like this for the samples exposed to background concentrations 0.01 M, 0.1 M and 1.0 M (profiles for 0.05 M and 0.4 M are not presented in Vl07)
The chloride concentration increases near the interfaces in all samples; we have discussed this interface excess effect in previousposts. Vl07 deal with this issue by evaluating the averages only for the inner parts of the samples. I performed a similar evaluation, also presented in the above figures (blue lines). In this evaluation I adopted the criterion to exclude all points situated less than 2 mm from the interfaces (Vl07 seem to have chosen points a bit differently). The clay concentration reevaluated in this way is also listed in the above table (last column). Given that I have only used nominal density for each sample (I don’t have information on the actual density of the sample sections), I’d say that the re-evaluated values agree well with those de-derived from reported \(\epsilon_\mathrm{eff}\). One exception is the sample 1.9/0.01, which is seen to have concentration points all over the place (or maybe detection limit is reached?). While Vl07 choose the lowest three points in their evaluation, here we choose to discard this result altogether. I mean that it is rather clear that this concentration profile cannot be considered to represent equilibrium.
As the reevaluation gives similar values as those reported, and since
we lack information for a full analysis, we will use the values
de-derived from reported \(\epsilon_\mathrm{eff}\) in the continued
assessment (except for sample 1.9/0.01).
Diffusion related estimations
Vl07 determine diffusion parameters by fitting various mathematical expressions to flux data.4 Parameters fitted in this way generally depend on the underlying adopted model, and we have discussed how equilibrium concentrations can be extracted from such parameters in an earlier blog post. In Vl07 it is clear that the adopted mathematical and conceptual model is the effective porosity diffusion model. When first presented in the article, however, it is done so in terms of a sorption distribution coefficient (\(R_d\)) that is claimed to take on negative values for anions. The presented mathematical expressions therefore contain a so-called rock capacity factor, \(\alpha\), which relates to \(R_d\) as \(\alpha = \phi + \rho_d\cdot R_d\). But such use of a rock capacity factor is a mix-up of incompatible models that I have criticized earlier. However, in Vl07 the description involving a sorption coefficient is in words only — \(R_d\) is never brought up again — and all results are reported, interpreted and discussed in terms of effective (or “chloride-accessible”) porosity, labeled \(\epsilon\) or \(\epsilon_\mathrm{Cl}\). We here exclusively use the label \(\epsilon_\mathrm{eff}\) when referring to formulas in Vl07. The mathematics is of course the same regardless if we call the parameter \(\alpha\), \(\epsilon\), \(\epsilon_\mathrm{Cl}\), or \(\epsilon_\mathrm{eff}\).
Mass balance in the out-diffusion stage
Vl07 measured the amount of tracers accumulated in the two reservoirs during the out-diffusion stage. The flux into the left side reservoir, which served as source reservoir during the preceding through-diffusion stage, was completely obscured by significant amounts of tracers present in the confining filter, and will not be considered further (also Vl07 abandon this flux in their analysis). But the total amount of tracers accumulated in the right side reservoir, \(N_\mathrm{right}\),5 can be used to directly estimate the chloride equilibrium concentration.
The initial concentration profile in the out-diffusion stage is linear (it is the steady-state profile), and the total amount of tracers, \(N_\mathrm{tot}\),6 can be expressed
where \(\bar{c}_0\) is the initial clay concentration at the left side interface, and \(V_\mathrm{sample}\) (\(\approx\) 5 cm3) is the sample volume.
A neat feature of the out-diffusion process is that two thirds of the
tracers end up in the left side reservoir, and one third in the right
side reservoir, as illustrated in this simulation
\(\bar{c}_0\) can thus be estimated by using
\(N_\mathrm{tot} = 3\cdot N_\mathrm{right}\) in eq. 3, giving
where \(c_\mathrm{source}\) is the tracer concentration in the left side reservoir in the through-diffusion stage.7 Although eq. 4 depends on a particular solution to the diffusion equation, it is independent of diffusivity (the diffusivity in the above simulation is \(1\cdot 10^{-10}\) m2/s). Eq. 4 can in this sense be said to be a direct estimation of \(\bar{c}_0\) (from measured \(N_\mathrm{right}\)), although maybe not as “direct” as the measurement of stable chloride, discussed previously.
Vl07 state eq. 4 in terms of a “Cl-accessible porosity”, but this is still just an interpretation of the clay concentration; \(\bar{c}_0\) is, in contrast to \(\epsilon_\mathrm{eff}\), directly accessible experimentally in principle. From the reported values of \(\epsilon_\mathrm{eff}\) we may back-calculate \(\bar{c}_0\), using the relation \(\bar{c}_0 / c_\mathrm{source} = \epsilon_\mathrm{eff}/\phi\). Alternatively, we may use eq. 4 directly to evaluate \(\bar{c}_0\) from the reported values of \(N_\mathrm{right}\). Curiously, these two approaches result in slightly different values for \(\bar{c}_0/c_\mathrm{source}\). I don’t understand the cause for this difference, but since \(N_\mathrm{right}\) is what has actually been measured, we use these values to estimate \(\bar{c}_0.\) The resulting equilibrium concentrations are
Test
\(N_\mathrm{right}\) (10-10 mol)
\(\bar{c}_0/c_\mathrm{source}\) (-)
1.3/0.01
4.10
0.038
1.3/0.05
10.2
0.097
1.3/0.1
17.8
0.168
1.3/0.4
41.4
0.395
1.3/1.0
52.4
0.445
1.6/0.01
1.21
0.014
1.6/0.05
3.64
0.043
1.6/0.1
6.15
0.072
1.6/0.4
13.0
0.154
1.6/1.0
21.6
0.225
1.9/0.01
0.41
0.006
1.9/0.05
1.14
0.018
1.9/0.1
1.64
0.025
1.9/0.4
3.19
0.051
1.9/1.0
8.19
0.113
We have now investigated two independent estimations of the chloride equilibrium concentrations: from mass balance of chloride tracers in the out-diffusion stage, and from measured stable chloride content. Here are plots comparing these two estimations
The similarity is quite extraordinary! With the exception of two
samples (1.3/0.4 and 1.9/0.1), the equilibrium chloride concentrations
evaluated in these two very different ways are essentially the
same. This result strongly confirms that the evaluations are adequate.
Steady-state fluxes
Vl07 present the flux evolution in the through-diffusion stage only for a single test (1.6/1.0), and it looks like this (left diagram)
The outflux reaches a relatively stable value after about 7 days,
after which it is meticulously monitored for a quite long time period.
The stable flux is not completely constant, but decreases slightly
during the course of the test. We anyway refer to this part as the
steady-state phase, and to the preceding part as the transient phase.
One reason that the steady-state is not completely stable is, reasonably, that the source reservoir concentration slowly decreases during the course of the test. The estimated drop from this effect, however, is only about one percent,8 while the recorded drop is substantially larger, about 7%. Vl07 do not comment on this perhaps unexpectedly large drop, but it may be caused e.g. by the ongoing conversion of the bentonite to a purer sodium state (see above).
Most of the analysis in Vl07 is based on anyway assigning a single
value to the steady-state flux. Judging from the above plot, Vl07 seem
to adopt the average value during the steady-state phase, and it is
clear that the assigned value is well constrained by the measurements
(the drop is a second order effect). The steady-state flux can
therefore be said to be directly measured in the through-diffusion
stage, rather than being obtained from fitting a certain model to
data.
Vl07 only implicitly consider the steady-state flux, in terms of a fitted “effective diffusivity” parameter, \(D_e\) (more on this in the next section). We can, however, “de-derive” the corresponding steady-state fluxes using \(j_\mathrm{ss} = D_e\cdot c_\mathrm{source}/L\), where \(L\) (= 0.01 m) is sample length. When comparing different tests it is convenient to use the normalized steady state flux \(\widetilde{j}_\mathrm{ss} = j_\mathrm{ss}/c_\mathrm{source}\), which then relates to \(D_e\) as \(\widetilde{j}_\mathrm{ss} = D_e/L\). Indeed, “effective diffusivity” is just a scaled version of the normalized steady-state flux, and it makes more sense to interpret it as such (\(D_e\) is not a diffusion coefficient). From the reported values of \(D_e\) we obtain the following normalized steady-state fluxes (my apologies for a really dull table)
Test
\(D_e\) (10-12 m2/s)
\(\widetilde{j}_\mathrm{ss}\) (10-10 m/s)
1.3/0.01
2.6
2.6
1.3/0.05
7.5
7.5
1.3/0.1
16
16
1.3/0.4
25
25
1.3/1.0
49
49
1.6/0.01
0.39
0.39
1.6/0.05
1.1
1.1
1.6/0.1
2.3
2.3
1.6/0.4
4.6
4.6
1.6/1.0
10
10
1.9/0.01
0.033
0.033
1.9/0.05
0.12
0.12
1.9/0.1
0.24
0.24
1.9/0.4
0.5
0.5
1.9/1.0
1.2
1.2
Plotting \(\widetilde{j}_\mathrm{ss}\) as a function of background concentration gives the following picture
The steady-state flux show a very consistent behavior: for all three
densities, \(\widetilde{j}_\mathrm{ss}\) increases with background
concentration, with a higher slope for the three lowest background
concentrations, and a smaller slope for the two highest background
concentrations. Although we have only been able to investigate the
1.6/1.0 test in detail, this consistency confirms that the
steady-state flux has been reliably determined in all tests.
Transient phase evaluations
So far, we have considered estimations based on more or less direct
measurements: stable chloride concentration profiles, tracer mass
balance in the out-diffusion stage, and steady-state fluxes. A major
part of the analysis in Vl07, however, is based on fitting solutions
of the diffusion equation to the recorded flux.
Vl07 state somewhat different descriptions for the through- and
out-diffusion stages. For out-diffusion they use an expression for the
flux into the right side reservoir (the sample is assumed located
between \(x=0\) and \(x=L\))
where \(j_\mathrm{ss}\) is the steady-state flux,9 \(D_e\) is “effective diffusivity”, and \(\epsilon_\mathrm{eff}\) is the effective porosity parameter (Vl07 also state a similar expression for the diffusion into the left side reservoir, but these results are discarded, as discussed earlier). For through-diffusion, Vl07 instead utilize the expression for the amount tracer accumulated in the right side reservoir
were \(S\) denotes the cross section area of the sample.
It is clear that Vl07 use \(D_e\) and \(\epsilon_\mathrm{eff}\) as fitting parameters, but not exactly how the fitting was conducted. \(D_e\) seems to have been determined solely from the the through-diffusion data, while separate values are evaluated for \(\epsilon_\mathrm{eff}\) from the through- and out-diffusion stages. As already discussed, Vl07 also provide a third estimation of \(\epsilon_\mathrm{eff}\), based on mass-balance in the out-diffusion stage. To me, the study thereby gives the incorrect impression of providing a whole set of independent estimations of \(\epsilon_\mathrm{eff}\). Although eqs. 5 and 6 are fitted to different data, they describe diffusion in one and the same sample, and an adequate fitting procedure should provide a consistent, single set of fitted parameters \((D_e, \epsilon_\mathrm{eff})\). Even more obvious is that the estimation of \(\epsilon_\mathrm{eff}\) from fitting eq. 5 should agree with the estimation from the mass-balance in the out diffusion stage — the accumulated amount in the right side reservoir is, after all, given by the integral of eq. 5. A significant variation of the reported fitting parameters for the same sample would thus signify internal inconsistency (experimental- or modelwise).
In the following reevaluation we streamline the description by solely using fluxes as model expressions,4 and by emphasizing steady-state flux as a parameter, which I think gives particularly neat expressions,10 (“TD” and “OD” denote through- and out-diffusion, respectively)
Here we use the pore diffusivity, \(D_p\), instead of the combination \(D_e/\epsilon_\mathrm{eff}\) in the exponential factors, and \(\widetilde{j} = j/c_\mathrm{source}\) denotes normalized flux. This formulation clearly shows that the time evolution is governed solely by \(D_p\), and that \(\widetilde{j}_\mathrm{ss}\) simply acts as a scaling factor.
In my opinion, using \(\widetilde{j}_\mathrm{ss}\) and \(D_p\) gives a formulation more directly related to measurable quantities; the steady-state flux is directly accessible experimentally, as we just examined, and \(D_p\) is an actual diffusion coefficient (in contrast to \(D_e\)) that can be directly evaluated from clay concentration profiles. Of course, eqs. 7 and 8 provide the same basic description as eqs. 5 and 6, and \(\widetilde{j}_\mathrm{ss}\) and \(D_p\) are related to the parameters reported in Vl07 as
When reevaluating the reported data we focus on the above discussed consistency aspect, i.e. whether or not a single model (a single pair of parameters) can be satisfactory fitted to all available data for the same sample. In this regard, we begin by noting that the fitting parameters are already constrained by the direct estimations. We have already concluded that the recorded steady-state flux basically determines \(\widetilde{j}_\mathrm{ss}\), and if we combine this with the estimated chloride clay concentration, \(D_p\) is determined from \(j_\mathrm{ss} = \phi\cdot D_p\cdot \bar{c}_0/L\), i.e.
Here are plotted values of \(D_p\) evaluated in this manner
Note that these values basically remain constant for samples of similar density (within a factor of 2) as the background concentration is varied by two orders of magnitude. This is the expected behavior of an actual diffusion coefficient,11 and confirms the adequacy of the evaluation; the numerical values also compares rather well with corresponding values for “MX-80” bentonite, measured in closed-cell tests (indicated by dashed lines in the figure).
Using eq. 10, we can also evaluate values of \(D_p\) corresponding to
the various reported fitted parameters \(\epsilon_\mathrm{eff}\). The
result looks like this (compared with the above evaluations from
direct estimations)
As pointed out above, a consistent evaluation requires that the
parameters fitted to the out-diffusion flux (red) are very similar
to those evaluated from considering the mass balance in the same process
(blue). We note that the resemblance is quite reasonable, although
some values — e.g. tests 1.3/1.0 and 1.6/1.0 — deviate in a perhaps
unacceptable way.
\(D_p\) evaluated from reported through-diffusion parameters, on the other hand, shows significant scattering (green). As the rest of the values are considerably more collected, and as the steady-state fluxes show no sign whatsoever that the diffusion coefficient varies in such erratic manner, it is quite clear that this scattering indicates problems with the fitting procedure for the through-diffusion data.
The 1.6/1.0 test
To further investigate the fitting procedures, we take a detailed look at the 1.6/1.0 test, for which flux data is provided. Vl07 report fitted parameters \(D_e = 1.0\cdot 10^{-11}\) m2/s and \(\epsilon_\mathrm{eff} = 0.063\) to the through-diffusion data, corresponding to \(\widetilde{j}_\mathrm{ss} = 1.0\cdot 10^{-9}\) m/s and \(D_p = 1.6\cdot 10^{-10}\) m2/s. We have already concluded that the steady-state flux is well captured by this data, but to see how well fitted \(\epsilon_\mathrm{eff}\) (or \(D_p\)) is, lets zoom in on the transient phase
This diagram also contains models (eq. 7) with different values of \(D_p\), and with a slightly different value of \(j_\mathrm{ss}\).12 It is clear that the model presented in the paper (black) completely misses the transient phase, and that a much better fit is achieved with \(D_p = 9.7\cdot10^{-11}\) m2/s (and \(\widetilde{j}_\mathrm{ss} = 1.06\cdot 10^{-9}\) m/s) (red). This difference cannot be attributed to uncertainty in the parameter \(D_p\) — the reported fit is simply of inferior quality. With that said, we note that all information on the transient phase is contained within the first three or four flux points; the reliability could probably have been improved by measuring more frequently in the initial stage.13
A reason for the inferior fit may be that Vl07 have focused only on the linear part of eq. 6; the paper spends half a paragraph discussing how the approximation of this expression for large \(t\) can be used to extract the fitting parameters using linear regression. Does this mean that only experimental data for large times where used to evaluate \(D_e\) and \(\epsilon_\mathrm{eff}\)? Since we are not told how fitting was performed, we cannot answer this question. Under any circumstance, the evidently low quality of the fit puts in question all the reported \(\epsilon_\mathrm{eff}\) values fitted to through-diffusion data. This is actually good news, as several of the corresponding \(D_p\) values were seen to be incompatible with constraints from direct estimations. We can thus conclude with some confidence that the inconsistency conveyed by the differently evaluated fitting parameters does not indicate experimental shortcomings, but stems from bad fitting of the through-diffusion model. Therefore, we simply dismiss the reported \(\epsilon_\mathrm{eff}\) values evaluated in this way. Note that the re-fitted value for \(D_p\) \((9.7\cdot10^{-11}\) m2/s) is consistent with those evaluated from direct estimations.
We note that when fitting the transient phase, it is appropriate to
use a value of \(\widetilde{j}_\mathrm{ss}\) slightly larger than the
average value adopted by Vl07 (as the model does not account for the
observed slight drop of the steady-state flux). This is only a minor
variation in the \(\widetilde{j}_\mathrm{ss}\) parameter itself (from
\(1.02\cdot10^{-9}\) to \(1.06\cdot10^{-9}\) m/s), but, since this value
sets the overall scale, it indirectly influences the fitted value of
\(D_p\) (model fitting is subtle!).
More questions arise regarding the fitting procedures when also examining the presented out-diffusion stage for the 1.6/1.0 sample. The tabulated fitted value for this stage is \(\epsilon_\mathrm{eff}\) = 0.075, while it is implied that the same value has been used for \(D_e\) as evaluated from the the through-diffusion stage (\(1.0\cdot 10^{-11}\) m2/s). The corresponding pore diffusivity is \(D_p = 1.33\cdot 10^{-10}\) m2/s. The provided plot, however, contains a different model than tabulated, and looks similar to this one (left diagram)
Here the presented model (black dashed line) instead corresponds to \(D_p = 8.5\cdot 10^{-11}\) m2/s (or \(\epsilon_\mathrm{eff}\) = 0.118). The model corresponding to the tabulated value (orange) does not fit the data! I guess this error may just be due to a typo in the table, but it nevertheless gives more reasons to not trust the reported \(\epsilon_\mathrm{eff}\) values fitted to diffusion data.
The above diagram also shows the model corresponding to the reported parameters from the through-diffusion stage (black solid line). Not surprisingly, this model does not fit the out-diffusion data, confirming that it does not appropriately describe the current sample. The model we re-fitted in the through-diffusion stage (red), on the other hand, captures the outflux data quite well. By also slightly adjusting \(\widetilde{j}_{ss}\), from from \(1.06\cdot10^{-9}\) to \(0.99\cdot10^{-9}\) m/s, to account for the drop in steady-state flux during the course of the through-diffusion test, and by plotting in a lin-lin rather than a log-log diagram, the picture looks even better! In a lin-lin plot (right diagram), it is easier to note that the model presented in the graph of Vl07 actually misses several of the data points. Could it be that Vl07 used visual inspection of the model in a log-log diagram to assess fitting quality? If so, data points corresponding to very low fluxes are given unreasonably high weight.14 This could be (another) reason for the noted difference between \(D_p\) evaluated from fitted parameters to the out-diffusion flux, and from the total accumulated amount of tracer (which should be equal).
From examining the reported results of sample 1.6/1.0 we have seen that the fitting procedures adopted in Vl07 appear inappropriate, but also that a consistent model can be successfully fitted to all available data (using a single \(D_p\)). Vl07 don’t provide flux data for any other sample, but we must conclude that the reported fitted \(\epsilon_\mathrm{eff}\) parameters cannot be trusted. Luckily, the preformed refitting exercise confirms the results obtained from analysis of stable chloride profiles and accumulated amount of tracers in out-diffusion, and we conclude that these results most probably are reliable. The corresponding value of \(\bar{c}_0/c_\mathrm{source}\) (using eq. 11) for the refitted model is here compared with the estimations from direct measurements
Summary and verdict
Chloride equilibrium concentrations evaluated from mass balance of the tracer in the out-diffusion stage and from stable chloride content show remarkable agreement. On the other hand, the scattering of estimated concentrations increases substantially if they are also evaluated from the reported fitted diffusion parameters. This could indicate underlying experimental problems, as a consistent evaluation should result in a single value for the equilibrium concentration; the various evaluations — stable chloride, out-diffusion mass balance, through-diffusion fitting and out-diffusion fitting — relate, after all, to a single sample.
By reexamining the evaluations we have found, however, that the problem is associated with how the fitting to diffusion data has been conducted (and presented), rather than indicating fundamental experimental issues. In the test that we have been able to examine in detail (1.6/1.0), we found that the reported models do not fit data, but also that it is possible to satisfactorily refit a single model that is also compatible with the direct methods for evaluating the equilibrium concentration. For the rest of the samples, we have also been able to discard the fitted diffusion parameters, as they are not compatible e.g. with how the steady-state flux (very consistently) vary with density and background concentration.
For these reasons, we discard the reported “effective porosity”
parameters evaluated from fitting solutions of the diffusion equation
to flux data, and keep the results from direct measurements of
chloride equilibrium concentrations (from stable chloride profile
analysis and mass-balance in the out-diffusion stage). I judge the
resulting chloride equilibrium concentrations as reliable and that
they can be used for increased qualitative process understanding. I
furthermore judge the directly measured steady-state fluxes as
reliable. This study thus provide adequate values for both chloride
equilibrium concentrations and diffusion coefficients.
However, a frustrating problem is that, although the equilibrium concentrations are well determined, we have little information on the exact state of the samples in which they have been measured. We basically have to rely on that the “KWK” material is “similar” to “MX-80”, keeping in mind that “MX-80” is not really a uniform material (from a scientific point of view). Also, the exchangeable mono/divalent cation ratio is most probably quite different in samples contacted with different background concentrations.
Yet, I judge the present study to provide the best information
available on chloride equilibrium in compacted bentonite, and will use
it e.g. for investigating the salt exclusion mechanism in these
systems (Ialreadyhave). That this information is the best available is, however, also
a strong argument for that more and better constrained data is
urgently needed.
The (reliable) results are presented in the diagram below, which includes “confidence areas”, that takes into account the spread in equilibrium concentrations, in samples where more than a single evaluation were performed, and the estimated uncertainty in effective montmorillonite dry density (the actual points are plotted at nominal density, assuming 80% montmorillonite content)
[1] Vejsada et al. (2006) call their material “KWK 20-80”. In other contexts, I have also found the versions “KWK food grade” and “KWK krystal klear”. I have given up my attempts at trying to understand the difference between these “KWK” variants.
[3] This should be relatively straightforward, but I get at bit nervous e.g. about the presence of a rather arbitrary factor 0.85 in the presented formula (eq. 19 in Van Loon et al. (2007)).
[4] As always for these types of diffusion tests, the raw data consists of simultaneously measured values of time (\(\{t_i\}\)) and reservoir concentrations (\(\{c_i\}\)). From these, flux can be evaluated as (\(A\) is sample cross sectional area, and \(V_\mathrm{res}\) is reservoir volume)
\(\bar{j}_i\) is the mean flux in the time interval between \(t_{i-1}\)
and \(t_i\), and should be associated with the average time of the
same interval: \(\bar{t}_i = (t_i + t_{i-1})/2\). The above formula
assumes no solution replacement after the \((i-1)\):th measurement (if
the solution is replaced, \(\left (c_i – c_{i-1} \right )\) should be
replaced with \(c_i\)).
Alternatively one can work with the accumulated amount of substance, which e.g. is \(N(t_i) = \sum_{j=1}^i c_j\cdot V_\mathrm{res}\), in case the solution is replaced after each measurement. I prefer using the flux because eq. * only depends on two consecutive measurements, while \(N(t_i)\) in principle depends on all measurements up to time \(t_i\). Also, I think it is easier to judge how well e.g. a certain model fits or is constrained by data when using fluxes; the steady-state, for example, then corresponds to a constant value.
Van Loon et al. (2007) seem to have utilized both fluxes and accumulated amount of substance in their evaluations, as discussed in later sections.
[8] From total test time, recorded flux, and sample cross sectional area, we estimate that about \(5.8\cdot 10^{-8}\) mol of tracer is transferred from the source reservoir during the course of the test (\(50\) days\(\cdot 2.7\cdot 10^{-11}\) mol/m2/s\(\cdot 0.0005\) m2). This is about 1% of the total amount tracer, \(c_\mathrm{source} \cdot V_\mathrm{source} = 2.65 \cdot 10^{-5}\) M \(\cdot 0.2\) L = \(5.3\cdot 10^{-6}\) mol.
[9] Van Loon et al. (2007) label this parameter \(J_L\), and don’t relate it explicitly to the steady-state flux. From the experimental set-up it is clear, however, that the initial value of the out-diffusion flux (into the right side reservoir) is the same as the previously maintained steady-state flux. Note that the expressions for the fluxes in the out-diffusion stage in Van Loon et al. (2007) has the wrong sign.
[10] The description provided by eqs. 5 and 6 not only mixes expressions for flux and accumulated amount tracer, but also contains three dependent parameters \(D_e\), \(\epsilon_\mathrm{eff}\), and \(j_\mathrm{ss}\) (e.g. \(j_\mathrm{ss} = D_e/(c_\mathrm{source}\cdot L)\)). In this reformulation, the model parameters are strictly only \(\widetilde{j}_\mathrm{ss}\) and \(D_p\). We have also divided out \(c_\mathrm{source}\) to obtain equations for normalized fluxes. Note that the expression for \(\widetilde{j}_{TD}(L,t)\) is essentially the same that we have used in previousassessments of through-diffusion tests. Note also that eqs. 7 and 8 imply the relation \(\widetilde{j}_{OD}(L,t) = \widetilde{j}_{ss} – \widetilde{j}_{TD}(L,t)\), reflecting that the out-diffusion process is essentially the through-diffusion process in reverse.
[11] Note the similarity with that diffusivity also is basically independent of background concentration for simple cations. Note also that there is no reason to expect completely constant \(D_p\) for a given density, because the samples are not identically prepared (being saturated with saline solutions of different concentration).
[12] As we here consider a single sample, we alternate a bit sloppily between steady-state flux (\(j_\mathrm{ss} \)) and normalized steady-state flux (\(\widetilde{j}_\mathrm{ss}\)), but these are simply related by a constant: \(\widetilde{j}_\mathrm{ss} = j_\mathrm{ss} / c_\mathrm{source}\). For the 1.6/1.0 test this constant is (as tabulated) \(c_\mathrm{source} = 2.65\cdot 10^{-2}\) mol/m3.
[13] I think it is a bit amusing that the pattern of data points suggests measurements being performed on Mondays, Wednesdays, and Fridays (with the test started on a Wednesday).
[14] I have warned about the dangers of log-log plots earlier.
Mo03 performed both chloride and iodide through-diffusion tests on
“MX-80” bentonite, but here we focus on the chloride
results. However, since the only example in the paper of an outflux
evolution and corresponding concentration profile is for iodide, this
particular result will also be investigated. The tests were performed
at background concentrations of 0.01 M or 0.1 M NaClO4, and nominal
sample densities of 0.4, 0.8, 1.2, 1.6, and 1.8 g/cm3. We refer to a
single test by stating “nominal density/background concentration”,
e.g. a test performed at nominal density 1.6 and background
concentration 0.1 M is referred to as “1.6/0.1”.
Uncertainty of samples
The material used is discussed only briefly, and the only reference given for its properties is (Müller-Von Moos and Kahr, 1983). I don’t find any reason to believe that the “MX-80” batch used in this study actually is the one investigated in this reference, and have to assume the same type of uncertainty regarding the material as we did in the assessment of Muurinen et al (1988). I therefore refer to that blog post for a discussion on uncertainty in montmorillonite content, cation population, and soluble calcium minerals.
Density
The samples in Mo03 are cylindrical with radius 0.5 cm and length 0.5
cm, giving a volume of 0.39 cm3. This is quite small, and corresponds
e.g. only to about 4% of the sample size used in
Muurinen et al
(1988). With such a small volume, the samples are at the
limit for being considered as a homogeneous material, especially for
the lowest densities: the samples of density 0.4 g/cm3 contain 0.157 g
dry substance in total, while a single 1 mm3 accessory grain weighs
about 0.002 — 0.003 g.
Furthermore, as the samples are sectioned after termination, the
amount substance in each piece may be very small. This could cause
additional problems, e.g. enhancing the effect of drying. The
reported profile (1.6/0.1, iodide diffusion) has 10 sections in the
first 2 mm. As the total mass dry substance in this sample is 0.628 g,
these sections have about 0.025 g dry substance each (corresponding to
the mass of about ten 1 mm3 grains). For the lowest density, a similar
sectioning corresponds to slices of dry mass 0.006 g (the paper does
not give any information on how the low density samples were
sectioned).
Mo03 only report nominal densities for the samples, but from the above considerations it is clear that a substantial (but unknown) variation may be expected in densities and concentrations.
A common feature of many through-diffusion studies is that the sample
density appears to decrease in the first few millimeters near the
confining filters. We saw this effect in the profiles of
Muurinen et al (1988),
and it has been the topic of some
studies,
including Mo03. Here, we don’t consider any possible cause, but simply
note that the samples seem to show this feature quite generally (below
we discuss how Mo03 handle this). Since the samples of Mo03 are only
of length 5 mm, we may expect that the major part of them are affected
by this effect. Of course, this increases the uncertainty of the
actual density of the used samples.
Uncertainty of external solutions
Mo03 do not describe how the external solutions were prepared, other
than that they used high grade chemicals. We assume here that the
preparation did not introduce any significant uncertainty.
Since “MX-80” contains a substantial amount of divalent ions, connecting this material with (initially) pure sodium solutions inevitably initiates cation exchange processes. The extent of this exchange depends on details such as solution concentrations, reservoir volumes, number of solution replacements, time, etc…
Very little information is given on the volume of the external solution
reservoirs. It is only hinted that the outlet reservoir may be 25 ml,
and for the inlet reservoir the only information is
The volume of the inlet reservoir was sufficient to keep the concentration nearly constant (within a few percent) throughout the experiments.
Consequently, we do not have enough information to assess the exact ion population during the course of the tests. We can, however, simulate this process of “unintentional exchange” to get some appreciation for the amount of divalent ions still left in the sample, as we did in the assessment of Muurinen et al. (1988). Here are the results from calculating the exchange equilibrium between a sample initially containing 30% exchangeable charge in form of calcium (70% sodium), and external NaClO4 solutions of various concentrations and volumes
In these calculations we assume a sample of density 1.6 g/cm3 (except
when indicated), a volume of 0.39 cm3, a cation exchange capacity of
0.75 eq/kg, and a Ca/Na selectivity coefficient of 5.
These simulations make it clear that the tests performed at 0.01 M
most probably contain most of the divalent ions initially present in
the “MX-80” material: even with an external solution volume of 1000
ml, or with density 0.4 g/cm3, exchange is quite
limited. For the tests performed at 0.1 M we expect some exchange of
the divalent ions, but we really can’t tell to what extent, as the
exact value strongly depends on handling (solution volumes, if
solutions were replaced, etc.). That the exact ion population is
unknown, and that the divalent/monovalent ratio probably is different
for different samples, are obviously major problems of the study (the
same problems were identified
in Muurinen et al
(1988)).
Uncertainty of diffusion parameters
Diffusion model
Mo03 determine diffusion parameters by fitting a model to all
available data, i.e the outflux evolution and the concentration
profile across the sample at termination. The model is solved by a
numerical code (“ANADIFF”) that takes into account transport both in
clay samples and filters. The fitted parameters are an apparent
diffusivity, \(D_a\), and a so-called “capacity factor”,
\(\alpha\). \(\alpha\) is vaguely interpreted as being the combination of
a porosity factor \(\epsilon\), and a sorption distribution
coefficient \(K_d\), described as “a generic term devoid of mechanism”
It is claimed that for anions, \(K_d\) can be treated as negative, giving \(\alpha < \epsilon\). I have criticized this mixing of what actually are incompatible models in an earlier blog post. Strictly, this use of a “generic term devoid of mechanism” means that the evaluated \(\alpha\) should not be interpreted in any particular way. Nevertheless, the waythis study is referenced in otherpublications, \(\alpha\) is interpreted as an effective porosity. It should be noticed, however, that this study is performed with a background electrolyte of NaClO4. The only chloride (or iodide) present is therefore at trace level, and it cannot be excluded that a mechanism of true sorption influences the results (there are indications that this is the case in other studies).
For the present assessment we anyway assume that \(\alpha\) directly
quantifies the anion equilibrium between clay and the external
solution (i.e. equivalent to
the
incorrect way of
assuming that \(\alpha\) quantifies a volume accessible to
chloride). It should be kept in mind, though, that effects of anion
equilibrium and potential true sorption is not resolved by the
single parameter \(\alpha\).
where \(c\) is the concentration in the clay of the isotope under
consideration, and the diffusion coefficient is written \(D_p\) to
acknowledge that it is a pore diffusivity (when referring to models
and parameter evaluations in Mo03 we will use the notation
“\(D_a\)”). The boundary conditions are
Oddly, Mo03 model the system as if two independent diffusion processes are simultaneously active. They refer to these as the “fast” and the “slow” processes, and hypothesize that they relate to diffusion in interlayer water2 and “interparticle water”,3 respectively.
The “fast” process is the “ordinary” process that is assumed to reach steady state during the course of the test, and that is the focus of other through-diffusion studies. The “slow” process, on the other hand, is introduced to account for the frequent observation that measured tracer profiles are usually significantly non-linear near the interface to the source reservoir (discussed briefly above). I guess that the reason for this concentration variation is due to swelling when the sample is unloaded. But even if the reason is not fully clear, it can be directly ruled out that it is the effect of a second, independent, diffusion process — because this is not how diffusion works!
If anions move both in interlayers and “interparticle water”, they reasonably transfer back and forth between these domains, resulting in a single diffusion process (the diffusivity of such a process depends on the diffusivity of the individual domains and their geometrical configuration). To instead treat diffusion in each domain as independent means that these processes are assumed to occur without transfer between the domains, i.e. that the bentonite is supposed to contain isolated “interlayer pipes”, and “interparticle pipes”, that don’t interact. It should be obvious that this is not a reasonable assumption. Incidentally, this is how all multi-porous models assume diffusion to occur (while simultaneously assuming that the domains are in local equilibrium…).
We will thus focus on the “fast” process in this assessment, although we also use the information provided by the parameters for the “slow” process. Mo03 report the fitted values for \(D_a\) and \(\alpha\) in a table (and diagrams), and only show a comparison between model and measured data in a single case: for iodide diffusion at 0.1 M background concentration and density 1.6 g/cm3. To make any kind of assessment of the quality of these estimations we therefore have to focus on this experiment (the article states that these results are “typical high clay density data”).
Outflux
The first thing to note is that the modeled accumulated diffusive substance does not correspond to the analytical solution for the diffusion process. Here is a figure of the experimental data and the reported model (as presented in the article), that also include the solution to eqs. 1 and 2.
In fact, the model presented in Mo03 has an incorrect time dependency in the early stages. Here is a comparison between the presented model and analytical solutions in the transient stage
With the given boundary conditions, the solutions to the diffusion
equation inevitably has zero slope at \(t = 0\),4 reflecting
that it takes a finite amount of time for any substance to reach the
outflux boundary. The models presented in Mo03, on the other hand, has
a non-zero slope in this limit. I cannot understand the reason for
this (is it an underlying problem with the model, or just a graphical
error?), but it certainly puts all reported parameter values in doubt.
The preferred way to evaluate diffusion data is, in my opinion, to look
at the flux evolution rather than the evolution of the accumulated
amount of diffused substance. Converting the reported data to flux,
gives the following picture.5
From a flux evolution it is easier to establish the steady-state, as it reaches a constant. It furthermore gives a better understanding for how well constrained the model is by the data. As is seen from the figure, the model is not at all very well constrained, as the experimental data almost completely miss the transient stage. (And, again, it is seen that the model in the paper with \(D_a= 9\cdot 10^{-11}\) m/s2 does not correspond to the analytical solution.)
The short transient stage is a consequence of using thin samples (0.5 cm). Compared e.g. to Muurinen et al (1988), who used three times as long samples, the breakthrough time is here expected to be \(3^2 = 9\) times shorter. As Muurinen et al. (1988) evaluated breakthrough times in the range 1 — 9 days, we here expect very short times. Here are the breakthrough times for all chloride diffusion tests, evaluated from the reported diffusion coefficients (“fast” process) using the formula \(t_\mathrm{bt} = L^2/(6D_a)\).
Test
\(D_a\)
\(t_\mathrm{bt}\)
(m2/s)
(days)
0.4/0.01
\(8\cdot 10^{-10}\)
0.06
0.4/0.1
\(9\cdot 10^{-10}\)
0.05
0.4/0.1
\(8\cdot 10^{-10}\)
0.06
0.8/0.01
\(3.5\cdot 10^{-10}\)
0.14
0.8/0.1
\(3.5\cdot 10^{-10}\)
0.14
0.8/0.1
\(3.7\cdot 10^{-10}\)
0.13
1.2/0.01
\(1.4\cdot 10^{-10}\)
0.34
1.2/0.1
\(2.3\cdot 10^{-10}\)
0.21
1.2/0.1
\(2.0\cdot 10^{-10}\)
0.24
1.6/0.1
\(1.0\cdot 10^{-10}\)
0.48
1.8/0.01
\(2\cdot 10^{-11}\)
2.41
1.8/0.1
\(5\cdot 10^{-11}\)
0.96
1.8/0.1
\(5.5\cdot 10^{-11}\)
0.88
The breakthrough time is much shorter than a day in almost all tests! To sample the transient stage properly requires a sampling frequency higher than \(1/t_{bt}\). As seen from the provided example of a outflux evolution, this is not the case: The second measurement is done after about 1 day, while the breakthrough time is about 0.5 days (moreover, the first measurement appears as an outlier). We have no information on sampling frequency in the other tests, but note that to properly sample e.g. the tests at 0.8 g/cm3 requires measurements at least every third hour or so. For 0.4 g/cm3, the required sample frequency is once an hour! This design choice puts more doubt on the quality of the evaluated parameters.
Concentration profile
The measured concentration profile across the 1.6/0.1 iodide sample,
and corresponding model results are presented in Mo03 in a figure very
similar to this
Here the two models correspond to the “slow” and “fast” process discussed above (a division, remember, that don’t make sense). Zooming in on the “linear” part of the profile, we can compare the “fast” process with analytical solutions (eqs. 1 and 2)
The analytical solutions correspond directly to the outflux curves presented above. We note that the analytical solution with \(D_p = 9\cdot 10^{-11}\) m/s2 corresponds almost exactly to the model presented by Mo03. As this model basically has the same steady state flux and diffusion coefficient, we expect this similarity. It is, however, still a bit surprising, since the corresponding outflux curve of the model in Mo03 was seen to not correspond to the analytical solution. This continues to cast doubt on the model used for evaluating the parameters.
We furthermore note that the evolution of the activity of the source
reservoir is not reported. Once in the text is mentioned that the
“carrier concentration” is \(10^{-6}\) M, but since we don’t know how
much of this concentration corresponds to the radioactive isotope, we
can not directly compare with reported concentration profile across
the sample (whose concentration unit is counts per minute per cm3).
By extrapolating the above model curve with \(\alpha = 0.15\), we can
however deduce that the corresponding source activity for this
particular sample is \(C_0 = 1.26\cdot 10^5/0.15\) cpu/cm3
\(= 8.40\cdot 10^5\) cpu/cm3. But it is unsatisfying that we cannot
check this independently. Also, we can of course not assume that this
value of \(C_0\) is the same in any other of the tests (in particular
those involving chloride). We thus lack vital information (\(C_0\)) to
be able to make a full assessment of the model fitting.
It should furthermore be noticed that the experimental concentration profile does not constrain the models very well. Indeed, the adopted model (diffusivity \(9\cdot 10^{-11}\) m/s2) misses the two rightmost concentration points (which correspond to half the sample!). A model that fits this part of the profile has a considerable higher diffusivity, and a correspondingly lower \(\alpha\) (note that the product \(D_p\cdot \alpha\) is constrained by the steady-state flux, eq. 3).
More peculiarities of the modeling is found if looking at the “slow”
process (remember that this is not a real diffusion process!). Zooming
in on the interface part of the profile and comparing with analytical
solutions gives this picture
Here we note that an analytical solution coincides with the model presented in Mo03 with parameters \(D_a = 6\cdot 10^{-14}\) m2/s and \(\alpha = 1.12\) only if it is propagated for about 15 days! Given that no outflux measurements seem to have been performed after about 4 days (see above), I don’t now what to make of this. Was the test actually conducted for 15 days? If so, why is not more of the outflux measured/reported? (And why were the samples then designed to give a breakthrough time of only a few hours?)
Without knowledge of for how long the tests were conducted, the reported diffusion parameters becomes rather arbitrary, especially for the low density samples. For e.g. the samples of density 0.4 g/cm3, even the “slow” process has a diffusivity high enough to reach steady-state within a few days. Simulating the processes with the reported parameters gives the following profiles if evaluated after 1 and 4 days, respectively
The line denoted “total” is what should resemble the measured
(unreported) data. It should be clear from these plots that the
division of the profile into two separate parts is quite arbitrary. It
follows that the evaluated diffusion parameters for the process of
which we are interested (“fast”) has little value.
Summary and verdict
We have seen that the reported model fitting leaves a lot of unanswered questions: some of the model curves don’t correspond to the analytical solutions, information on evolution times and source concentrations is missing, and the modeled profiles are divided quite arbitrary into two separate contributions (which are not two independent diffusion process).
Moreover, the ion population (divalent vs. monovalent cations) of the samples are not known, but there are strong reasons to believe that the 0.01 M tests contain a significant amount of divalent ions, while the 0.1 M samples are partly converted to a more pure sodium state.
Also, the small size of the samples contributes to more uncertainty,
both in terms of density, but also for the flux evolution because the
breakthrough times becomes very short.
Based on all of these uncertainties, I mean that the results of Mo03
does not contribute to quantitative process understanding and my
decision is to not to use the study for e.g. validating models
of anion exclusion.
A confirmation of the uncertainty in this study is given by
considering the density dependence on the chloride equilibrium
concentrations for constant background concentration, evaluated from
the reported diffusion parameters (\(\alpha\) for the “fast” process).
If these results should be taken at face value, we have to accept a
very intricate density dependence: for 0.1 M background, the
equilibrium concentration is mainly constant between densities 0.3
g/cm3 and 0.7 g/cm3, and increases
between densities 1.0 g/cm3 and 1.45 g/cm3 (or,
at least, does not decrease). For 0.01 M background, the equilibrium
concentration instead falls quite dramatically between between
densities 0.3 g/cm3 and 0.7 g/cm3, and
thereafter displays only a minor density dependence.
To accept such dependencies, I require a considerably more rigorous experimental procedure and evaluation. In this case, I rather view the above plot as a confirmation of large uncertainties in parameter evaluation and sample properties.
[1] Strictly, \(c(0,t)\) relates to the concentration in the endpoint of the inlet filter. But we ignore filter resistance in this assessment, which is valid for the 1.6/0.1 sample. Moreover, the filter diffusivities are not reported in Mo03.
[2] Mo03 refer to interlayer pores as
“intralayer” pores, which may cause some confusion.
[3] Apparently, the authors assume an
underlying
stack view of the material.
[4] It may be
objected that the analytical solution do not include the filter
resistance. But note that filter resistance only will increase the
delay. Moreover, the transport capacity of the sample in this test
is so low that filters have no significant influence.
[5] The model by Mo03 looks noisy
because I have read off values of accumulated concentration from the
published graph. The “noise” occurs because the flux is evaluated
from the concentration data by the difference formula:
where \(t_i\) and \(t_{i+1}\) are the time coordinates for two consequitive data points, \(a(t)\) is the accumulated amount diffused substance at time \(t\), \(A\) is the cross sectional area of the sample, \(\bar{t}_i = (t_{i+1} + t_i)/2\) is the average time of the considered time interval, and \(\bar{j}\) denotes the average flux during this time interval.
where \(\phi\) is the porosity of the sample, \(D_c\) is the macroscopic
pore diffusivity of the presumed interlayer domain, and \(\Xi\) is the
ion equilibrium coefficient. \(\Xi\) quantifies the ratio between
internal and external concentrations of the ion under consideration,
when the two compartments are in equilibrium.
where \(\epsilon_\mathrm{eff}\) is the porosity of a presumed bulk water
domain where anions are assumed to reside exclusively, and \(D_p\) is
the corresponding pore diffusivity of this bulk water domain.
We have
discussed earlier
how the homogeneous mixture and the effective porosity models can be
equally well fitted to a specific set of anion through-diffusion
data. The parameter “translation” is simply
\(\phi\cdot \Xi \leftrightarrow \epsilon_\mathrm{eff}\) and
\(D_c \leftrightarrow D_p\). It may appear from this equivalency that
diffusion data alone cannot be used to discriminate between the two
models.
But note that the interpretation of how \(D_e\) varies with background
concentration is very different in the two models.
In the homogeneous mixture model, \(D_c\) is not expected to vary with background concentration to any greater extent, because the diffusing domain remains essentially the same. \(D_e\) varies in this model primarily because \(\Xi\) varies with background concentration, as a consequence of an altered Donnan potential.
In the effective porosity model, \(D_p\) is expected to vary, because the volume of the bulk water domain, and hence the entire domain configuration (the “microstructure”), is postulated to vary with background concentration. \(D_e\) thus varies in this model both because \(D_p\) and \(\epsilon_\mathrm{eff}\) varies.
A simple way of taking into account a varying domain configuration (as in the effective porosity model) is to assume that \(D_p\) is proportional to \(\epsilon_\mathrm{eff}\) raised to some power \(n – 1\), where \(n > 1\). Eq. 2 can then be written
where \(D_0\) is the tracer diffusivity in pure bulk water. Eq. 3 is in the bentonite literature often referred to as “Archie’s law”, in analogy with a similar evaluation in more conventional porous systems. Note that with \(D_0\) appearing in eq. 3, this expression has the correct asymptotic behavior: in the limit of unit porosity, the effective diffusivity reduces to that of a pure bulk water domain.
Eq. 3 shows that \(D_e\) in the effective porosity model is expected to depend non-linearly on background concentration for constant sample density. In contrast, since \(D_c\) is not expected to vary significantly with background concentration, we expect a linear dependence of \(D_e\) in the homogeneous mixture model. Keeping in mind the parameter “translation” \(\phi\cdot\Xi \leftrightarrow \epsilon_\mathrm{eff}\), the prediction of the homogeneous mixture model (eq. 1) can be expressed1
We have thus managed to establish a testable difference between the effective porosity and the homogeneous mixture model (eqs. 3 and 4). This is is great! Making this comparison gives us a chance to increase our process understanding.
Comparison with experiment
Van Loon et al. (2007)
It turns out that the chloride diffusion measurements performed by Van Loon et al. (2007) are accurate enough to resolve whether \(D_e\) depends on “\(\epsilon_\mathrm{eff}\)” according to eqs. 3 or 4. As will be seen below, this data shows that \(D_e\) varies in accordance with the homogeneous mixture model (eq. 4). But, since Van Loon et al. (2007) themselves conclude that \(D_e\) obeys Archie’s law, and hence complies with the effective porosity model, it may be appropriate to begin with some background information.
Van Loon et al. (2007) report three different series of diffusion tests, performed on bentonite samples of density 1300, 1600, and 1900 kg/m3, respectively. For each density, tests were performed at five different NaCl background concentrations: 0.01 M, 0.05 M, 0.1 M, 0.4 M, and 1.0 M. The tests were evaluated by fitting the effective porosity model, giving the effective diffusion coefficient \(D_e\) and corresponding “effective porosity” \(\epsilon_\mathrm{eff}\) (it is worth repeating that the latter parameter equally well can be interpreted in terms of an ion equilibrium coefficient).
Van Loon et al. (2007) conclude that their data complies with eq. 3, with \(n = 1.9\), and provide a figure very similar to this one
Here are compared evaluated values of effective diffusivity and “effective porosity” in various tests. The test series conducted by Van Loon et al. (2007) themselves are labeled with the corresponding sample density, and the literature data is from García-Gutiérrez et al. (2006)2 (“Garcia 2006”) and the PhD thesis of A. Muurinen (“Muurinen 1994”). Also plotted is Archie’s law with \(n\) =1.9. The resemblance between data and model may seem convincing, but let’s take a further look.
Rather than lumping together a whole bunch of data sets, let’s focus on the three test series from Van Loon et al. (2007) themselves, as these have been conducted with constant density, while only varying background concentration. This data is thus ideal for the comparison we are interested in (we’ll get back to commenting on the other studies).
It may also be noted that the published plot contains more data points (for these specific test series) than are reported in the rest of the article. Let’s therefore instead plot only the tabulated data.3 The result looks like this
Here we have also added the predictions from the homogeneous mixture model (eq. 4), where \(D_c\) has been fitted to each series of constant density.
The impression of this plot is quite different from the previous one: it should be clear that the data of Van Loon et al. (2007) agrees fairly well with the homogeneous mixture model, rather than obeying Archie’s law. Consequently, in contrast to what is stated in it, this study refutes the effective porosity model.
The way the data is plotted in the article is reminiscent of Simpson’s paradox: mixing different types of dependencies of \(D_e\) gives the illusion of a model dependence that really isn’t there. Reasonably, this incorrect inference is reinforced by using a log-log diagram (I have warned about log-log plots earlier). With linear axes, the plots give the following impression
This and the previous figure show that \(D_e\) depends approximately linearly on “\(\epsilon_\mathrm{eff}\)”, with a slope dependent on sample density. With this insight, we may go back and comment on the other data points in the original diagram.
García-Gutiérrez et al. (2006) and Muurinen et al. (1988)
The tests by García-Gutiérrez et al. (2006) don’t vary the background concentration (it is not fully clear what the background concentration even is4), and each data point corresponds to a different density. This data therefore does not provide a test for discriminating between the models here discussed.
I have had no access to Muurinen (1994), but by examining the data, it is clear that it originates from Muurinen et al. (1988), which was assessed in detail in a previous blog post. This study provides two estimations of “\(\epsilon_\mathrm{eff}\)”, based on either breakthrough time or on the actual measurement of the final state concentration profile. In the above figure is plotted the average of these two estimations.5
One of the test series in Muurinen et al. (1988) considers variation of density while keeping background concentration fixed, and does not provide a test for the models here discussed. The data for the other two test series is re-plotted here, with linear axis scales, and with both estimations for “\(\epsilon_\mathrm{eff}\)”, rather than the average6
As discussed in the assessment of this study, I judge this data to be too uncertain to provide any qualitative support for hypothesis testing. I think this plot confirms this judgment.
Glaus et al. (2010)
The measurements by Van Loon et al. (2007) are enough to convince me that the dependence of \(D_e\) for chloride on background concentration is furtherevidence for that a homogeneous view of compacted bentonite is principally correct. However, after the publication of this study, the same authors (partly) published more data on chloride equilibrium, in pure Na-montmorillonite and “Na-illite”,7 in Glaus et al. (2010).
This data certainly shows a non-linear relation between \(D_e\) and “\(\epsilon_\mathrm{eff}\)” for Na-montmorillonite, and Glaus et al. (2010) continue with an interpretation using “Archie’s law”. Here I write “Archie’s law” with quotation marks, because they managed to fit the expression to data only by also varying the prefactor. The expression called “Archie’s law” in Glaus et al. (2010) is
where \(A\) is now a fitting parameter. With \(A\) deviating from \(D_0\), this expression no longer has the correct asymptotic behavior as expected when interpreting \(\epsilon_\mathrm{eff}\) as quantifying a bulk water domain (see eq. 3). Nevertheless, Glaus et al. (2010) fit this expression to their measurements, and the results look like this (with linear axes)
Here is also plotted the prediction of the homogeneous mixture model
(eq. 4). For the montmorillonite data, the dependence is
clearly non-linear, while for the “Na-illite” I would say that the
jury is still out.
Although the data for montmorillonite in
Glaus et al. (2010)
is
non-linear, there are several strong arguments for why this is not an
indication that the effective porosity model is correct:
Remember that this result is not a confirmation of the measurements in Van Loon et al. (2007). As demonstrated above, those measurements complies with the homogeneous mixture model. But even if accepting the conclusion made in that publication (that Archie’s law is valid), the Glaus et al. (2010) results do not obey Archie’s law (but “Archie’s law”).
The four data points correspond to background concentrations of 0.1 M, 0.5 M, 1.0 M, and 2.0 M. If “\(\epsilon_\mathrm{eff}\)” represented the volume of a bulk water phase, it is expected that this value should level off, e.g. as the Debye screening length becomes small (Van Loon et al. (2007) argue for this). Here “\(\epsilon_\mathrm{eff}\)” is seen to grow significantly, also in the transition between 1.0 M and 2.0 M background concentration.
These are Na-montmorillonite samples of dry density 1.9 g/cm3. With an “effective porosity” of 0.067 (the 2.0 M value), we have to accept more than 20% “free water” in these very dense systems! This is not even accepted by otherproponents of bulk water in compacted bentonite.
Furthermore, these tests were performed with a background of \(\mathrm{NaClO_4}\), in contrast to Van Loon et al. (2007), who used chloride also for the background. The only chloride around is thus at trace level, and I put my bet on that the observed non-linearity stems from sorption of chloride on some system component.
Insight from closed-cell tests
Note that the issue whether or not \(D_e\) varies linearly with
“\(\epsilon_\mathrm{eff}\)” at constant sample density is equivalent
to whether or not \(D_p\) (or \(D_c\)) depends on background
concentration. This is similar to how presumed concentration
dependencies of the pore diffusivity for simple cations
(“apparent”
diffusivities) have been used to argue for multi-porosity in compacted
bentonite. For cations,
a closer look shows that no such dependency is found in the
literature.
For anions, it is a bit frustrating that the literature data is not
accurate or relevant enough to fully settle this issue (the data of
Van Loon et al. (2007)
is, in my opinion, the best available).
However, to discard the conceptual view underlying the effective porosity model, we can simply use results from closed-cell diffusion studies. In Na-montmorillonite equilibrated with deionized water, Kozaki et al. (1998) measured a chloride diffusivity of \(1.8\cdot 10^{-11}\) m2/s at dry density 1.8 g/cm3.8 If the effective porosity hypothesis was true, we’d expect a minimal value for the diffusion coefficient9 in this system, since \(\epsilon_\mathrm{eff}\) approaches zero in the limit of vanishing ionic strength. Instead, this value is comparable to what we can evaluate from e.g. Glaus et al. (2010) at 1.9 cm3/g, and 2.0 M background electrolyte: \(D_e/\epsilon_\mathrm{eff} = 7.2\cdot 10^{-13}/0.067\) m2/s = \(1.1\cdot 10^{-11}\) m2/s.
That chloride diffuses just fine in dense montmorillonite equilibrated with pure water is really the only argument needed to debunk the effective porosity hypothesis.
Footnotes
[1] Note that \(\epsilon_\mathrm{eff}\) is not a parameter in the homogeneous mixture model, so eq. 4 looks a bit odd. But it expresses \(D_e\) if \(\phi\cdot \Xi\) is interpreted as an effective porosity.
[3] This choice is not critical for the conclusions made in this blog post, but it seems appropriate to only include the data points that are fully described and reported in the article.
[4] García-Gutiérrez et al. (2004) (which is the study compiled in García-Gutiérrez et al. (2006)) state that the samples were saturated with deionized water, and that the electric conductivity in the external solution were in the range 1 — 3 mS/cm.
[5] The data point labeled with a “?” seems to have been obtained by making this average on the numbers 0.5 and 0.08, rather than the correctly reported values 0.05 and 0.08 (for the test at nominal density 1.8 g/cm3 and background concentration 1.0 M).
[6] Admittedly, also the data we have plotted from the original tests in Van Loon et al. (2007) represents averages of several estimations of “\(\epsilon_\mathrm{eff}\)”. We will get back to the quality of this data in a future blog post when assessing this study in detail, but it is quite clear that the estimation based on the direct measurement of stable chloride is the more robust (it is independent of transport aspects). Using these values for “\(\epsilon_\mathrm{eff}\)”, the corresponding plot looks like this
[7] To my mind, it is a misnomer to describe something as illite in sodium form. Although “illite” seems to be a bit vaguely defined, it is clear that it is supposed to only contain potassium as counter-ion (and that these ions are non-exchangeable; the basal spacing is \(\sim\)10 Å independent of water conditions). The material used in Glaus et al. (2010) (and severalotherstudies) has a stated cation exchange capacity of 0.22 eq/kg, which in a sense is comparable to the montmorillonite material (a factor 1/4). Shouldn’t it be more appropriate to call this material e.g. “mixed-layer”?
[8] This value is the average from two tests performed at 25 °C. The data from this study is better compiled in Kozaki et al. (2001).
[9] Here we refer of course to the empirically defined diffusion coefficient, which I have named \(D_\mathrm{macr.}\) in earlier posts. This quantity is model independent, but it is clear that it should be be associated with the pore diffusivities in the two models here discussed (i.e. with \(D_c\) in the homogeneous mixture model, and with \(D_p\) in the effective porosity model).
Mu88 performed both chloride and uranium through-diffusion tests on “MX-80” bentonite, as well as sorption tests. Here we focus solely on the chloride diffusion. We also disregard one diffusion test series that does not vary external concentration (it was conducted with an unspecified “artificial groundwater” and varied sample density).
Left are two test series performed with nominal sample densities 1.2 g/cm3 and 1.8 g/cm3, respectively. For each of these densities, chloride through-diffusion tests were performed with external NaCl concentrations of 0.01 M, 0.1 M, and 1.0 M, respectively. The samples were cylindrical with a diameter of 3.0 cm, and a length of 1.5 cm, giving a volume of 10.6 cm3. To refer to a specific test or sample, we use the nomenclature “nominal density/external concentration”, e.g. the test performed at nominal density 1.2 g/cm3 and external solution 0.1 M is referred to as “1.2/0.1”.
Uncertainty of bentonite samples
“MX-80” is not the name of some specific standardized material, but simply a product name.2 It is quite peculiar that that “MX-80” nevertheless is a de facto standard in the research field for clay buffers in radwaste repositories. But, being a de facto standard, several batches of bentonite with this name have been investigated and reported throughout the years. We consequently have some appreciation for its constitution, and the associated variation.
In Mu88, the material used is only mentioned by name, and it is only
mentioned once (in the abstract!). We therefore can’t tell which of
the studies that is more appropriate to refer to. Instead, let’s take
a look at how “MX-80” has been reported generally.
*) These values were derived from summing the exchangeable ions, and are probably overestimations.
Montmorillonite content
Reported montmorillonite content varies in the range 75 — 85%. For the present context, this primarily gives an uncertainty in adopted effective montmorillonite dry density, which, in turn, is important for making relevant comparison between bentonite materials with different montmorillonite content. For the “MX-80” used in Mu88 we here assume a montmorillonite content of 80%. In the table below is listed the corresponding effective montmorillonite densities when varying the montmorillonite content in the range \(x =\) 0.75 — 0.85, for the two nominal dry densities.
Dry density
EMDD (\(x\)=0.75)
EMDD (\(x\)=0.80)
EMDD (\(x\)=0.85)
(g/cm3)
(g/cm3)
(g/cm3)
(g/cm3)
1.2
1.01
1.05
1.09
1.8
1.61
1.66
1.70
The uncertainty in montmorillonite content thus translates to an
uncertainty in effective montmorillonite dry density on the order of
0.1 g/cm3.
Cation population
While reported values of the cation exchange capacity of “MX-80” are relatively constant, of around 0.75 eq/kg,4 the reported fraction of sodium ions is seen to vary, in the range 70 — 85 %. The remaining population is mainly di-valent rare-earth metal ions (calcium and magnesium). This does not only mean that different studies on “MX-80” may give results for quite different types of systems, as the mono- to di-valent ion ratio may vary, but also that samples within the study may represent quite different systems. We examine this uncertainty below, when discussing the external solutions.
Soluble calcium minerals
The uncertainty of how much divalent cations are available is in fact larger than just discussed. “MX-80” is reported to contain a certain amount of soluble calcium minerals, in particular gypsum. These provide additional sources for divalent ions, which certainly will be involved in the chemical equilibration as the samples are water saturated. Reported values of gypsum content in “MX-80” are on the order of 1%. With a molar mass of 0.172 kg/mol, this contributes to the calcium content by \(2\cdot 0.01/0.172\) eq/kg \(\approx 0.12\) eq/kg, or about 16% of the cation exchange capacity.
Sample density
The samples in Mu88 that we focus on have nominal dry density of 1.2
and 1.8 g/cm3. The paper also reports measured porosities on each
individual sample, listed in the below table together with
corresponding values of dry density5
Test
\(\phi\)
\(\rho_d\)
(-)
(g/cm3)
1.2/0.01
0.54
1.27
1.2/0.1
0.52
1.32
1.2/1.0
0.49
1.40
1.8/0.01
0.37
1.73
1.8/0.1
0.31
1.89
1.8/1.0
0.34
1.81
We note a substantial variation in measured density for samples with the same nominal density: for the 1.2 g/cm3 samples, the standard deviation is 0.06 g/cm3, and for the 1.8 g/cm3 samples it is 0.07 g/cm3. Moreover, while the mean value for the 1.8 g/cm3 samples is close to the nominal value (1.81 g/cm3), that for the 1.2 g/cm3 samples is substantially higher (1.33 g/cm3).
It is impossible to know from the information provided in Mu88 if this
uncertainty is intrinsic to the procedure of preparing the samples, or
if it is more related to the procedure of measuring the density at
test termination.6
Uncertainty of external solutions
Mu88 do not describe how the external solutions were prepared. We
assume here, however, that preparing pure NaCl solutions gives no
significant uncertainty.
Further, the paper contains no information on how the samples were water saturated, nor on the external solution volumes. Since samples with an appreciable amount of di-valent cations are contacted with pure sodium solutions, it is unavoidable that an ion exchange process is initiated. As we don’t know any detail of the preparation process, this introduces an uncertainty of the exact aqueous chemistry during the course of a test.
To illustrate this problem, here are the results from calculating the
exchange equilibrium between a sample initially containing 30%
exchangeable charge in form of calcium (70% sodium), and external
NaCl solutions of various concentrations and volumes
In these calculations we assume a sample of density 1.8 g/cm3 with the
same volume as in Mu88 (10.6 cm3), a cation exchange capacity of 0.75
eq/kg, and a Ca/Na selectivity coefficient of 5.
In a main series, we varied the external volume between 50 and 1000 ml
(solid lines). While the solution volume naturally has a significant
influence on the process, it is seen that the initial calcium content
essentially remain for the lowest concentration (0.01 M). In contrast,
for a 1.0 M solution, a significant amount of calcium is exchanged for
all the solution volumes.
The figure also shows a case for sample density 1.2 g/cm3 (dashed line), and a scenario where equilibrium has been obtained twice, with a replacement of the first solution (to a once again pure NaCl solution) (dot-dashed line).
The main lesson from these simulations is that the actual amount of di-valent ions present during a diffusion test depends on many details: the way samples were saturated, volume of external solutions, if and how often solutions were replaced, time, etc. It is therefore impossible to state the exact ion population in any of the tests in Mu88. But, guided by the simulations, it seems very probable that the tests performed at 0.01 M contain a substantial amount of di-valent ions, while those performed at 1.0 M probably resemble more pure sodium systems.
The only information on external solutions in Mu88 is that the
“solution on the low concentration side was changed regularly”
during the course of a test. This implies that the amount of di-valent
cations may not even be constant during the tests.
Uncertainty of diffusion parameters
The diffusion parameters explicitly listed in Mu88 are \(D_e\) and “\(D_a\)”, while it is implicitly understood that they have been obtained by fitting the effective porosity model to outflux data and the measured clay concentration profile in the final state. “\(D_a\)” is thus really the pore diffusivity \(D_p\),7 and relates to \(D_e\) as \(D_e = \epsilon_\mathrm{eff} D_p\), where \(\epsilon_\mathrm{eff}\) is the so-called “effective porosity”. In a previous blog post, we discussed in detail how anion equilibrium concentrations can be extracted from through-diffusion tests, and the results derived there is used extensively in this section.
Rather than fitting the model to the full set of data (i.e. outflux
evolution and final state concentration profile), diffusion parameters
in Mu88 have been extracted in various limits.
Evaluation of \(D_e\) in Mu88
The effective diffusivity was obtained by estimating the steady-state flux, dividing by external concentration difference of the tracer, and multiplying by sample length \begin{equation} D_e = \frac{j^\mathrm{ss}\cdot L}{c^\mathrm{source}}\tag{1} \end{equation}
Here it is assumed that the target reservoir tracer concentration can
be neglected (we assume this
throughout). Eq. 1 is basically eq. 1 in
Mu88
(and
eq. 8 in the earlier blog post), from which we can evaluate the
values of the steady-state flux that was used for the reported values
of \(D_e\) (\(A \approx 7.1\) cm2 denotes sample cross sectional area)
Test
\(D_e\)
\(A\cdot j^\mathrm{ss}/c^\mathrm{source}\)
(\(\mathrm{m^2/s}\))
(ml/day)
1.2/0.01
\(7.7\cdot 10^{-12}\)
0.031
1.2/0.1
\(2.9\cdot 10^{-11}\)
0.118
1.2/1.0
\(1.2\cdot 10^{-10}\)
0.489
1.8/0.01
\(3.3\cdot 10^{-13}\)
0.001
1.8/0.1
\(4.8\cdot 10^{-13}\)
0.002
1.8/1.0
\(4.0\cdot 10^{-12}\)
0.016
The figure below compares the evaluated values of the steady-state
flux with the flux evaluated from the measured target concentration
evolution,8 for samples with nominal dry
density 1.8 g/cm3 (no concentration data was reported for the 1.2
g/cm3 samples)
These plots clearly show that the transition to steady-state is only
resolved properly for the test with highest background concentration
(1.0 M). It follows that the uncertainty of the evaluated steady-state
— and, consequently, of the evaluated \(D_e\) values — increases
dramatically with decreasing background concentration for these
samples.
Evaluation of \(D_p\) in Mu88
Pore diffusivities were obtained in two different ways. One method was to relate the steady-state flux to the clay concentration profile at the end of the test, giving \begin{equation} D_{p,c} = \frac{j^\mathrm{ss}\cdot L}{\phi\cdot\bar{c}(0)} \tag{2} \end{equation}
where \(\bar{c}(0)\) denotes the chloride clay concentration at the interface to the source reservoir. The quantity in eq. 2 is called “\(D_{ac}\)”7 in Mu88, and this equation is essentially the same as eq. 2 in Mu889 (and eq. 10 in the previous blog post). Using the steady-state fluxes, we can back-calculate the values of \(\bar{c}(0)\) used for this evaluation of \(D_{p,c}\)
Test
\(D_{p,c}\)
\(A\cdot j^\mathrm{ss}/c^\mathrm{source}\)
\(\phi\)
\(\bar{c}(0)/c^\mathrm{source}\)
(\(\mathrm{m^2/s}\))
(ml/day)
(-)
(-)
1.2/0.01
\(7.0\cdot 10^{-11}\)
0.031
0.54
0.204
1.2/0.1
\(2.8\cdot 10^{-10}\)
0.118
0.52
0.199
1.2/1.0
\(5.1\cdot 10^{-10}\)
0.489
0.49
0.480
1.8/0.01
\(2.0\cdot 10^{-11}\)
0.001
0.37
0.045
1.8/0.1
\(3.1\cdot 10^{-11}\)
0.002
0.31
0.050
1.8/1.0
\(5.2\cdot 10^{-11}\)
0.016
0.34
0.226
Note that, although we did some calculations to obtain them, the values for \(\bar{c}(0)/c^\mathrm{source}\) in this table are closer to the actual measured raw data (concentrations). We made the calculation above to “de-derive” these values from the reported diffusion coefficients (combining eqs. 1 and 2 shows that \(\bar{c}(0)\) is obtained from the reported parameters as \(\bar{c}(0)/c^\mathrm{source} = D_e/(\phi D_{p,c})\)).
Here are compared the measured concentration profiles for the samples
of nominal density 1.8 g/cm3 and the corresponding slopes used to
evaluate \(D_{p,c}\) (profiles for the 1.2 g/cm3 samples are not
provided in Mu88)
For background concentrations 1.0 M and 0.1 M, the evaluated slope corresponds quite well to the raw data. For the 0.01 M sample, however, the match is not very satisfactory. I suspect that a detection limit may have been reached for the analysis of the profile of this sample. Needless to say, the evaluated value of \(\bar{c}(0)\) is very uncertain for the 0.01 M sample.
It may also be noted that all measured concentration profiles deviates from linearity near the interface to the source reservoir. This is a general behavior in through-diffusion tests, which I am quite convinced of is related to sample swelling during dismantling, but there are also other suggestedexplanations. Here we neglect this effect and relate diffusion quantities to the linear parts of profiles, but this issue should certainly be treated in a separate discussion. Update (220407): non-linear profiles are discussed here.
\(D_p\) was also evaluated in a different way in Mu88, by measuring what we here will call the breakthrough time, \(t_\mathrm{bt}\) (Mu88 call it “time-lag”). This quantity is fairly abstract, and relates to the asymptotic behavior of the analytical expression for the outflux that apply for constant boundary concentrations (we here assume them to be \(c^\mathrm{source}\) and 0, respectively). This expression is displayed in eq. 7 in the previous blog post.
Multiplying the outflux by the sample cross sectional area \(A\) and integrating, gives the accumulated amount of diffused tracers. In the limit of long times, this quantity is, not surprisingly, linear in \(t\) \begin{equation} A\cdot j^\mathrm{ss} \cdot \left(t – \frac{L^2}{6\cdot D_p} \right ) \end{equation}
\(t_\mathrm{bt}\) is defined as the time for which this asymptotic
expression is zero. Determining \(t_\mathrm{bt}\) from the measured
outflux evolution consequently allows for an estimation of \(D_p\) as
\begin{equation}
D_{p,t} = \frac{L^2}{6t_\mathrm{bt}} \tag{3}
\end{equation}
This quantity is called “\(D_{at}\)” in Mu887
(eq. 3 is eq. 3 in Mu88). With another back
calculation we can extract the values of \(t_\mathrm{bt}\) determined
from the raw data
Test
\(D_{p,t}\)
\(t_\mathrm{bt}\)
(\(\mathrm{m^2/s}\))
(days)
1.2/0.01
\(1.4\cdot 10^{-10}\)
3.1
1.2/0.1
\(2.0\cdot 10^{-10}\)
2.2
1.2/1.0
\(3.2\cdot 10^{-10}\)
1.4
1.8/0.01
\(5.0\cdot 10^{-11}\)
8.7
1.8/0.1
\(5.4\cdot 10^{-11}\)
8.0
1.8/1.0
\(7.7\cdot 10^{-11}\)
5.6
These evaluated breakthrough times are indicated
in the flux plots above for samples of
nominal dry density 1.8 g/cm3. For the 0.1 M and 0.01 M
samples it is obvious that this value is very uncertain — without a
certain steady-state flux it is impossible to achieve a certain
breakthrough time. The breakthrough time for the 1.8/1.0 test, on
the other hand, simply appears to be incorrectly evaluated: in terms
of outflux vs. time, the breakthrough time should be the time where
the flux has reached 62% of the steady-state
value.10
As no raw data is reported for the 1.2 g/cm3 tests, the quality of the
evaluated breakthrough times cannot be checked for them. It may be
noted, however, that the evaluated breakthrough times are
significantly shorter in this case as compared with the 1.8 g/cm3
tests. Consequently, while the sampling frequency is high enough to
properly resolve the transient stage of the outflux evolution for the
1.8g/cm3 tests, it must be substantially higher in order to resolve
this stage in the 1.2g/cm3 tests (I guess a rule of thumb is that
sampling frequency must be at least higher than \(1/t_{bt}\)).
In a well conducted study these estimates should be similar; \(D_{p,c}\) and \(D_{p,t}\) are, after all, estimations of the same quantity: the pore diffusivity \(D_p\).7 But here we note a discrepancy of approximately a factor 2 between several values of \(\bar{c}(0)\).
It is difficult to judge generally which of the estimations are more
accurate, but we have seen that for the 1.8/0.1 and 1.8/0.01 tests,
the flux data is not very well resolved, giving a
corresponding uncertainty on the equilibrium concentration estimated
from the breakthrough time. On the other hand, also
the concentration profile is poorly
resolved in the case of 0.01 M at 1.8 g/cm3.
However, in cases where the value of \(\bar{c}(0)/c^\mathrm{source}\) is substantial (as for the 1.8/1.0 test and, reasonably, for all tests at 1.2 g/cm3), we expect the estimation directly from the concentration profile to be accurate and robust (as for the 1.8 g/cm3 test at high NaCl concentration). For the 1.2 g/cm3 samples we cannot say much more than this, since Mu88 don’t provide the concentration raw data. For the 1.8/1.0 test, however, we can continue the analysis by fitting the model to all available data.
Re-evaluation by fitting to the full data set
Note that all evaluations in Mu88 are based on making an initial estimation of the steady-state flux, giving \(D_e\) (eq. 1). This value of \(D_e\) (or \(j^{ss}\)) is thereafter fixed in the subsequent estimation of \(D_{p,c}\) (eq. 2). Likewise, an estimation of the steady-state flux is required for estimating the breakthrough time. Here is an animation showing the variation of the model when transitioning from the value of the pore diffusivity estimated from breakthrough time (\(7.7\cdot 10^{-11}\) m2/s), to the value estimated from concentration profile (\(5.2\cdot 10^{-11}\) m2/s) for the 1.8/1.0 test, keeping the steady-state flux fixed at the initial estimation
Note that the axes for the flux is on top (time) and to the right (accumulation rate). This animation confirms that the diffusivity evaluated from breakthrough time in Mu88 gives a way too fast process: the slope of the steady-state concentration profile is too small, and the outflux evolution has a too short transient stage. On the other hand, using the diffusivity estimated from the concentration profiles still doesn’t give a flux that fit very well. The problem is that this fitting is performed with a fixed value of the steady-state flux. By instead keeping the slope fixed at the experimental values, while varying diffusivity (and thus steady state flux), we get the following variation
This animation shows that the model can be fitted well to all data (at least for the 1.8/1.0 test). The problem with the evaluation in Mu88 is that it assumes the steady-state to be fully reached at the later stages of the test. As the above fitting procedure shows, this is only barely true. The experiments could thus have been designed better by conducting them longer, in order to better sample the steady-state phase (and the steady-state flux should have been fitted to the entire data set). Nevertheless, for this sample, the steady-state flux obtained by allowing for this parameter to vary is only slightly different from that used in Mu88 (17.5 rather than 16.3 \(\mathrm{\mu}\)l/day, corresponding to a change of \(D_p\) from \(5.2\cdot10^{-11}\) to \(5.6\cdot10^{-11}\) m2/s). Moreover, this consideration should not be a problem for the 1.2 g/cm3 tests, if they were conducted for as long time as the 1.8 g/cm3 tests, because steady-state is reached much faster (in those tests, sampling frequency may instead be a problem, as discussed above).
As we were able to fit the full model to all data, we conclude that the value of \(\bar{c}(0)/c^\mathrm{source}\) obtained from \(D_{p,c}\) is probably the more robust estimation11, and that there appears to be a problem with how the breakthrough times have been determined. For the 1.8 g/cm3 samples we have demonstrated that this is the case, for the 1.2 g/cm3 we can only make an educated guess that this is the case.
Summary and verdict
We have seen that the results on chloride diffusion in Mu88 suffer from uncertainty from several sources:
The “MX-80” material is not that well defined
Densities vary substantially for samples at the same nominal density
Without knowledge of e.g water saturation procedures and solution volumes, it is impossible to estimate the proper ion population during the course of a test
It is, however, highly likely that tests performed at low NaCl concentrations contain substantial amounts of di-valent ions, while those at high NaCl concentration are closer to being pure sodium systems.
The reported diffusivities give a corresponding uncertainty in the chloride equilibrium concentrations of about a factor of 2. While some tests essentially have a too high noise level to give certain estimations, the problem for the others seems to stem from the estimation of breakthrough times.
Here is an attempt to encapsulate the above information in an
updated plot for the chloride equilibrium data in Mu88
The colored squares represent “confidence areas” based on the variation within each nominal density (horizontally), and on the variation of \(\bar{c}(0)/c^\mathrm{source}\) from the two reported values on pore diffusivity7 (vertically). The limits of these rectangles are simply the 95% confidence interval, based on these variations, and assuming a normal distribution.
Data points put within parentheses are estimations judged to be
improper (based on either re-evaluation of the raw data, or informed
guesses).
From the present analysis my decision is to not use the data
from Mu88 to e.g. validate models for anion exclusion. Although there
seems to be nothing fundamentally wrong with how these test were
conducted, they suffer from so many uncertainties of various sources
that I judge the data to not contribute to quantitative process
understanding.
[2] MX-80 is not only a brand name, but also
a band
name.
[3] This report is “Bentonite Mineralogy” by L. Carlson (Posiva WR 2004-02), but it appears to not be included in the INIS database. It can, however, be found with some elementary web searching.
[4] It’s interesting to note that the cation exchange capacity of
“MX-80” remains more or less constant, while the montmorillonite
content has some variation. This implies that the montmorillonite
layer charge varies (and is negatively correlated with montmorillonite
content). Could it be that the manufacturer has a specified cation
exchange capacity as requirement for this product?
[5] To convert porosity to
dry density, I used \(\rho_d = \rho_s\cdot(1-\phi)\), with solid grain
density \(\rho_s = 2.75\) g/cm3.
[6] A speculation is that the uncertainty stems from the measurement procedure, as this was done on smaller sections of the full samples. It is not specified in Mu88 what the reported porosity represent, but it is reasonable to assume that it is the average of all sections of a sample.
[8] These values were not tabulated, but I have read
them off from the graphs in Mu88.
[9] Mu88 use the
concentration based on the total volume in their expression, while
\(\bar{c}\) is
defined in terms of water volume (water mass,
strictly). Eq.2 therefore contains the physical
porosity. In their concentration profile plots, however, Mu88 use
\(\bar{c}\) as variable (called \(c_{pw}\) — the “concentration in the pore
water”)
[10] Plugging the breakthrough time \(L^2/6D_p\) into the expression for the flux gives
I find it amusing that this value is close to the reciprocal golden ratio (0.618033…). Finding the breakthrough time from a flux vs. time plot thus corresponds (approximately) to splitting the y-axis according to the golden ratio.
[11] Note that the actual evaluated values of $D_{p,c}$ in Mu88 still may be uncertain, because they also depend on the values of the steady-state flux, which we have seen were not optimally evaluated.
On the surface, “Ionendiffusion in Hochverdichtetem Bentonit”1 by G. Kahr, R. Hasenpatt, and M. Müller-Vonmoos, published by NAGRA in March 1985, looks like an ordinary mundane 37-page technical report. But it contains experimental results that could have completely changed the history of model development for compacted clay.
Test principles
The tests were conducted in a quite original manner. By compacting
granules or powder, the investigators obtained samples that
schematically look like this
The bentonite material — which was either Na-dominated “MX-80”, or Ca-dominated “Montigel” — was conditioned to a specific water-to-solid mass ratio \(w\). At one of the faces, the bentonite was mixed with a salt (in solid form) to form a thin source for diffusing ions. This is essentially the full test set-up! Diffusion begins as soon as the samples are prepared, and a test was terminated after some prescribed amount of time, depending on diffusing ion and water content. At termination, the samples were sectioned and analyzed. In this way, the investigators obtained final state ion distributions, which in turn were related to the initial states by a model, giving the diffusion coefficients of interest.
Note that the experiments were conducted without exposing samples to a liquid (external) solution; the samples were “unsaturated” to various degree, and the diffusing ions dissolve within the bentonite. The samples were not even confined in a test cell, but “free-standing”, and consequently not under pressure. They were, however, stored in closed vessels during the course of the tests, to avoid changes in water content.
With this test principle a huge set of diffusion tests were
performed, with systematic variation of the following variables:
Bentonite material (“MX-80” or “Montigel”)
Water-to-solid mass ratio (7% — 33%)
Dry density (1.3 g/m3 — 2.1 g/m3 )
Diffusing salt (SrCl2, SrI2, CsCl, CsI, UO2(NO3)2, Th(NO3)4, KCl, KI, KNO3, K2SO4, K2CO3, KF)
Distribution of water in the samples
From e.g. X-ray diffraction (XRD) we know that bentonite water at low water content is distributed in distinct, sub-nm thin films. For simplicity we will refer to all water in the samples as interlayer water, although some of it, reasonably, forms interfaces with air. The relevant point is that the samples contain no bulk water phase, but only interfacial (interlayer) water.
I argueextensively on this blog for that interlayer water is the only relevant water phase also in saturated samples under pressure. In the present case, however, it is easier to prove that this is the case, as the samples are merely pressed bentonite powder at a certain water content; the bentonite water is not pressurized, the samples are not exposed to liquid bulk water, nor are they in equilibrium with liquid bulk water. Since the water in the samples obviously is mobile — as vapor, but most reasonably also in interconnected interlayers — it is a thermodynamic consequence that it distributes as to minimize the chemical potential.
There is a ton of literature on how the montmorillonite basal spacing
varies with water content. Here, we use the neat result from
Holmboe et al. (2012)
that the average interlayer distance varies basically
linearly2 with water content, like this
XRD-studies also show that bentonite water is distributed in rather distinct hydration states, corresponding to 0, 1, 2, or 3 monolayers of water.3 We label these states 0WL, 1WL, 2WL, and 3WL, respectively. In the figure is indicated the approximate basal distances for pure 1WL (12.4 Å), 2WL (15.7 Å), and 3WL (19.0 Å), which correspond roughly to water-to-solid mass ratios of 0.1, 0.2, and 0.3, respectively.
From the above plot, we estimate roughly that the driest samples in
Kahr et al. (1985)
(\(w \sim 0.1\)) are in pure 1WL states, then transitions to a mixture
of 1WL and 2WL states (\(w\sim 0.1 – 0.2\)), to pure 2WL states
(\(w \sim 0.2\)), to a mixture of 2WL and 3WL states
(\(w\sim 0.2 – 0.3\)), and finally to pure 3WL states (\(w\sim 0.3\)).
Results
With the knowledge of how water is distributed in the samples, let’s
take a look at the results of
Kahr et al. (1985).
Mobility of interlayer cations confirmed
The most remarkable results are of qualitative character. It is, for
instance, demonstrated that several cations diffuse far into the
samples. Since the samples only contain interlayer water, this is a
direct proof of ion mobility in the interlayers!
Also, cations are demonstrated to be mobile even when the water
content is as low as 7 or 10 %! As such samples are dominated by 1WL
states, this is consequently evidence for ion mobility in 1WL states.
A more quantitative assessment furthermore shows that the cation diffusivities varies with water content in an almost step-wise manner, corresponding neatly to the transitions between various hydration states. Here is the data for potassium and strontium
This behavior further confirms that the ions diffuse in interlayers,
with an increasing diffusivity as the interlayers widen.
It should also be noted that the evaluated values of the diffusivities
are comparable to — or even larger4 — than
corresponding results from saturated, pressurized tests.
This strongly suggests that interlayer diffusivity dominates also in
the latter types of tests, which also has been
confirmedin more recent years. The
larger implication is that interlayer diffusion is the only relevant
type of diffusion in general in compacted bentonite.
Anions enter interlayers (and are mobile)
The results also clearly demonstrate that anions (iodide) diffuse in systems with water-to-solid mass ratio as low as 7%! With no other water around, this demonstrates that anions diffuse in — and consequently have access to — interlayers. This finding is strongly confirmed by comparing the \(w\)-dependence of diffusivity for anions and cations. Here is plotted the data for iodide and potassium (with the potassium diffusivity indicated on the right y-axis)
The iodide mobility increases as the system transitions from 1WL to 2WL, in a very similar way as for potassium (and strontium). If this is not a proof that the anion diffuse in the same domain as the cation I don’t know what is! Also for iodide the value of the diffusivity is comparable to what is evaluated in water saturated systems under pressure, which implies that interlayer diffusivity dominates generally in compacted bentonite, also for anions.
Dependence of diffusivity on water content and density
A conclusion made in
Kahr et al. (1985),
that I am not sure I fully agree with, is that diffusivity mainly
depends on water content rather than density. As seen in the diagrams
above, the spread in diffusivity is quite substantial for a given
value of \(w\). There is actually some systematic variation here: for
constant \(w\), diffusivity tend to increase with dry density.
Although using unsaturated samples introduces additional variation, the present study provides a convenient procedure to study diffusion in systems with very low water content. A more conventional set-up in this density limit has to deal with enormous pressures (on the order of 100 MPa).
Interlayer chemistry
An additional result is not acknowledged in the report, but is a direct consequence of the observations: the tests demonstrate that interlayers are chemically active. The initially solid salt evidently dissolves before being able to diffuse. Since these samples are not even close to containing a bulk water phase (as discussed above), the dissolution process must occur in an interlayer. More precisely, the salt must dissolve in interface water between the salt mineral and individual montmorillonite layers, as illustrated here
This study seems to have made no impact at all
In the beginning of 1985, the research community devoted to radioactive waste barriers seems to have been on its way to correctly identify diffusion in interlayers as the main transport mechanism, and to recognize how ion diffusion in bentonite is influenced by equilibrium with external solutions.
Already in 1981,
Torstenfelt et al. (1981)
concluded that the
traditional diffusion-sorption model is not valid,
for e.g. diffusion of Sr and Cs, in compacted bentonite. They also
noted, seemingly without realizing the full importance, that these
ions diffused even in unsaturated samples with as low water-to-solid
mass ratio as 10%.
A significant diffusion was observed for Sr in dry clay, although slower than for water saturated clay, Figure 4, while Cs was almost immobile in the dry clay.
A year later also
Eriksen and Jacobsson (1982)
concluded that the traditional diffusion model is not valid. They
furthermore pointed out the subtleties involved when interpreting
through-diffusion experiments, due to ion equilibrium effects
One difficulty in correlating the diffusivities obtained from profile analysis to the diffusivities calculated from steady state transport data is the lack of knowledge of the tracer concentration at the solution-bentonite interface. This concentration is generally higher for sorbing species like positive ions (counterions to the bentonite) and lower for negative ions (coions to the bentonite) as shown schematically in figure 11. The equilibrium concentration of any ion in the bentonite and solution respectively is a function of the ionic charge, the ionic strength of the solution and the overall exchanger composition and thereby not readily calculated
By regarding the clay-gel as a concentrated electrolytic system Marinsky has calculated (30) distribution coefficients for Sr2+ and Cs+ ions in good agreement with experimentally determined Kd-values. The low anionic exchange capacity and hence the low anion concentration in the pore solution caused by Donnan exclusion also explain the low concentrations of anionic tracers within the clay-gel
[…]
For simple cations the ion-exchange process is dominating and there is, as also pointed out by Marinsky (30), no need to suppose that the counterions are immobilized. It ought to be emphasized that for the compacted bentonite used in the diffusion experiments discussed in this report the water content corresponds roughly to 2-4 water molecule layers (31). There is therefore really no “free water” and the measured diffusivity \(\bar{D}\) can be regarded as corresponding approximately to the diffusivity within the adsorbed phase […]
Furthermore, also
Soudek et al. (1984)
had discarded the traditional diffusion-sorption model, identified the
exchangeable cations as giving a dominating contribution to mass
transfer, and used Donnan equilibrium calculations to account for the
suppressed internal chloride concentration.
In light of this state of the research front, the contribution of Kahr et al. (1985) cannot be described as anything but optimal. In contrast to basically all earlier studies, this work provides systematic variation of several variables (most notably, the water-to-solid ratio). As a consequence, the results provide a profound confirmation of the view described by Eriksen and Jacobsson (1984) above, i.e. that interlayer pores essentially govern all physico-chemical behavior in compacted bentonite. A similar description was later given by Bucher and Müller-Vonmoos (1989) (though I don’t agree with all the detailed statements here)
There is no free pore water in highly compacted bentonite. The water in the interlayer space of montmorillonite has properties that are quite different from those of free pore water; this explains the extremely high swelling pressures that are generated. The water molecules in the interlayer space are less mobile than their free counterparts, and their dielectric constant is lower. The water and the exchangeable cations in the interlayer space can be compared to a concentrated salt solution. The sodium content of the interlayer water, at a water content of 25%, corresponds approximately to a 3-n salt solution, or six times the concentration in natural seawater. This more or less ordered water is fundamentally different from that which engineers usually take into account; in the latter case, pore water in a saturated soil is considered as a freely flowing fluid. References to the porosity in highly compacted bentonite are therefore misleading. Highly compacted bentonite is an unfamiliar material to the engineer.
Given this state of the research field in the mid-80s, I find it
remarkable that history took a different turn. It appears as the
results of
Kahr et al. (1985)
made no impact at all (it may be noticed that they themselves analyzed
the results in terms of the traditional diffusion-sorption
model). And rather than that researchers began identifying that
transport in interlayers is the only relevant contribution, the
so-called surface diffusion model gained popularity (it was already promoted by
e.g.
Soudek et al. (1984)
and
Neretnieks and Rasmuson (1983)). Although this
model emphasizes mobility of the exchangeable cations, it is still
centered around the idea that compacted bentonite contains bulk
water.5 Most
modern bentonite models
suffer from similar flaws: they are formulated in terms of bulk water,
while many effects related to interlayers are treated as irrelevant or
optional.
For the case of anion diffusion the historical evolution is maybe even more disheartening. In 1985 the notions of “effective” or “anion-accessible” porosities seem to not have been that widely spread, and here was clear-cut evidence of anions occupying interlayer pores. But just a few years later the idea began to grow that the pore space in compacted bentonite should be divided into regions which are either accessible or inaccessible to anions. As far as I am aware, the first use of the term “effective porosity” in this context was by Muurinen et al. (1988), who, ironically, seem to have misinterpreted the Donnan equilibrium approach presented by Soudek et al. (1984). To this day, this flawed concept is central in many descriptions of compacted clay.
Footnotes
[1] “Ion
diffusion in highly compacted bentonite”
[2] Incidentally, the slope of this line corresponds to a water “density” of 1.0 g/cm3.
[3] This is the region of swelling often
referred to as
“crystalline”.
[4] I’m not sure the evaluation in Kahr et al. (1985) is fully correct. They use the solution to the diffusion equation for an impulse source (a Gaussian), but, to my mind, the source is rather one of constant concentration (set by the solubility of the salt). Unless I have misunderstood, the mathematical expression to be fitted to data should then be an erfc-function, rather than a Gaussian. Although this modification would change the numerical values of the evaluated diffusion coefficients somewhat, it does not at all influence the qualitative insights provided by the study.
[5] I have discussed the surface diffusion model in some detail in previousblogposts.
Recently, we discussed reported equilibrium chloride concentrations in sodium dominated bentonite, and identified a need to assess the individual studies. As most data is obtained from through-diffusion experiments, we here take a general look at how anion equilibrium is a part of the through-diffusion set-up, and how we can use reported model parameters to extract the experimentally accessible equilibrium concentrations.
We define the experimentally accessible concentration of a chemical
species in a bentonite sample as
where \(n\) is the total amount of the
species,1 and \(m_{w}\) is
the total water mass in the
clay.2 It should be clear
that \(\bar{c}\), which we will refer to as the clay concentration, is
accessible without relying on any particular model concept.
An equilibrium concentration is defined as the corresponding clay concentration (i.e. \(\bar{c}\)) of a species when the clay is in equilibrium with an external solution with species concentration \(c^\mathrm{ext}\). A convenient way to express this equilibrium is in terms of the ratio \(\bar{c}/c^\mathrm{ext}\).
The through-diffusion set-up
A through-diffusion set-up consists of a (bentonite) sample sandwiched between a source and a target reservoir, as illustrated schematically here (for some arbitrary time):
The sample length is labeled \(L\), and we assume the sample to be
initially empty of the diffusing species. A test is started by adding
a suitable amount of the diffusing species to the source
reservoir. Diffusion through the bentonite is thereafter monitored by
recording the concentration evolution in the target
reservoir,3 giving
an estimation of the flux out of the sample (\(j^\mathrm{out}\)). The
clay concentration for anions is typically lower than the
corresponding concentration in the source reservoir.
Although a through-diffusion test is not in full equilibrium (by definition), local equilibrium prevails between clay and external solution4 at the interface to the source reservoir (\(x=0\)). Thus, even if the source concentration varies, we expect the ratio \(\bar{c}(0)/c^\mathrm{source}\) to stay constant during the course of the test.5
The effective porosity diffusion model
Our primary goal is to extract the concentration ratio \(\bar{c}(0)/c^\mathrm{source}\) from reported through-diffusion parameters. These parameters are in many anion studies specific to the “effective porosity” model, rather than being accessible directly from the experiments. We therefore need to examine this particular model.
The effective porosity model divides the pore space into a bulk water domain and a domain that is assumed inaccessible to anions. The porosity of the bulk water domain is often referred to as the “effective” or the “anion-accessible” porosity, and here we label it \(\epsilon_\mathrm{eff}\).
Anions are assumed to diffuse in the bulk water domain according to
Fick’s first law
where \(D_p\) is the pore diffusivity in the bulk water phase. This
relation is alternatively expressed as
\(j = -D_e \cdot \nabla c^\mathrm{bulk}\), which defines the effective
diffusivity \(D_e = \epsilon_\mathrm{eff} \cdot D_p\).
Diffusion is assumed to be the only mechanism altering the
concentration, leading to Fick’s second law
where \(\phi\) is the physical porosity of the sample. Since a bulk water concentration varies continuously across interfaces to external solutions, we have \(c^\mathrm{bulk}(0) = c^\mathrm{source}\) at the source reservoir, giving
This equation shows that the effective porosity parameter quantifies
the anion equilibrium concentration that we want to extract. That is
not to say that the model is valid (more on that later), but that we
can use eq. 4 to translate reported model parameters to an
experimentally accessible quantity.
In principle, we could finish the analysis here, and use eq. eq. 4 as our main result. But most researchers do not evaluate the effective porosity in the direct way suggested by this equation (they may not even measure \(\bar{c}\)). Instead, they evaluate \(\epsilon_\mathrm{eff}\) from a fitting procedure that also includes the diffusivity as a parameter. It is therefore fruitful to also include the transport aspects of the through-diffusion test in our analysis.
From closed-cell diffusion tests, we know that the clay concentration evolves according to Fick’s second law, both for many cations and anions. We will therefore take as an experimental fact that \(\bar{c}\) evolves according to
This equation defines the diffusion coefficient \(D_\mathrm{macr.}\), which should be understood as an empirical quantity.
Combining eqs. 3 and 2 shows that \(D_p\) governs the evolution of \(\bar{c}\) in the effective porosity model (if \(\epsilon_\mathrm{eff}/\phi\) can be considered a constant). A successful fit of the effective porosity model to experimental data thus provides an estimate of \(D_\mathrm{macr.}\) (cf. eq. 5), and we may write
With the additional assumption of constant reservoir concentrations,
eq. 2 has a relatively simple analytical solution, and the
corresponding outflux reads
where \(j^\mathrm{ss}\) is the steady-state flux. In steady-state,
\(c^\mathrm{bulk}\) is distributed linearly across the sample, and we
can express the gradient in eq. 1 using the reservoir
concentrations, giving
Treating \(j^\mathrm{ss}\) as an empirical parameter (it is certainly
accessible experimentally), and using eq. 6, we get another expression
for \(\epsilon_\mathrm{eff}\) in terms of experimentally accessible
quantities
This relation (together with eqs. 4 and 6) demonstrates that if we fit eq. 7 using \(D_p\) and \(j^\mathrm{ss}\) as fitting parameters, the equilibrium relation we seek is given by
This procedure may look almost magical, since any explicit reference to the effective porosity model has now disappeared; eq. 10 can be viewed as a relation involving only experimentally accessible quantities.
But the validity of eq. 10 reflects the empirical fact that the (steady-state) flux can be expressed using the gradient in \(\bar{c}\) and the physical porosity. The effective porosity model can be successfully fitted to anion through-diffusion data simply because it complies with this fact. Consequently, a successful fit does not validate the effective porosity concept, and essentially any description for which the flux can be expressed as \(j = -\phi\cdot D_p \cdot \nabla\bar{c}\) will be able to fit to the data.
We may thus consider a generic model for which eq. 5 is valid and for which a steady-state flux is related to the external concentration difference as
where \(\beta\) is an arbitrary constant. Fitting such a model, using \(\beta\) and \(D_p\) as parameters, will give an estimate of \(\bar{c}(0)/c^\mathrm{source}\) (\(=\beta / \phi\)).
Note that the system does not have to reach steady-state — eq. 11 only states how the model relates a steady-state flux to the reservoir concentrations. Moreover, the model being fitted is generally numerical (analytical solutions are rare), and may account for e.g. possible variation of concentrations in the reservoirs, or transport in the filters connecting the clay and the external solutions.
The effective porosity model emerges from this general description by interpreting \(\beta\) as quantifying the volume of a bulk water phase within the bentonite sample. But \(\beta\) can just as well be interpreted e.g. as an ion equilibrium coefficient (\(\phi\cdot \Xi = \beta\)), showing that this description also complies with the homogeneous mixture model.
Additional comments on the effective porosity model
The effective porosity model can usually be successfully fitted to anion through-diffusion data (that’s why it exists). The reason is not because the data behaves in a manner that is difficult to capture without assuming that anions are exclusively located in a bulk water domain, but simply because this model complies with eqs. 5 and 11. We have seen that also the homogeneous mixture model — which makes the very different choice of having no bulk water at all within the bentonite — will fit the data equally well: the two fitting exercises are equivalent, connected via the parameter identification \(\epsilon_\mathrm{eff} \leftrightarrow \phi\cdot\Xi\).
Perhaps even more remarkable is that authors frequently treat the effective porosity model as was it some version of the traditional diffusion-sorption model. This is often done by introducing a so-called rock capacity factor \(\alpha\) — which can take on the values \(\alpha = \phi + \rho\cdot K_d\) for cations, and \(\alpha = \epsilon_\mathrm{eff}\) for anions — and write \(D_e = \alpha D_a\), where \(D_a\) is the “apparent” diffusion coefficient. The reasoning seems to go something like this: since the parameter in the governing equation in one model can be written as \(D_e/\epsilon_\mathrm{eff}\), and as \(D_e/(\phi + \rho\cdot K_d)\) in the other, one can view \(\epsilon_\mathrm{eff}\) as being due to negative sorption (\(K_d < 0\)).
But such a mixing of completely different mechanisms (volume restriction vs. sorption) is just a parameter hack that throws most process understanding out the window! In particular, it hides the fact that the effective porosity and diffusion-sorption models are incompatible: their respective bulk water domains have different volumes. Furthermore, this lumping together of models has led to that anion diffusion coefficients routinely are reported as “apparent”, although they are not; the underlying model contains a pore diffusivity (eq. 2). As I have stated before, the term “apparent” is supposed to convey the meaning that what appears as pure diffusion is actually the combined result of diffusion, sorption, and immobilization. Sadly, in the bentonite literature, “apparent diffusivity” often means “actual diffusivity”.
Footnotes
[1] For anions, the total amount is relatively easy to measure by e.g. aqueous extraction. Cations, on the other hand, will stick to the clay, and need to be exchanged with some other type of cation (not initially present). In any case, the total amount of a species (\(n\)) can in principle be obtained experimentally, in an unambiguous manner.
[2] Another reasonable choice would be to divide by the total sample volume.
[3] If the test is designed as to have a significant change of the source concentration, it is a good idea to also measure the concentration evolution in this reservoir.
[5] Provided that the rest of the aqueous chemistry remains constant, which is not always the case. For instance, cation exchange may occur during the course of the test, if the set-up involves more than one type of cation, and there may be ongoing mineral dissolution.