In some of the simulations that we discussed in a previous blog post on molecular dynamics (MD) studies on anion exclusion, chloride never enters the montmorillonite interlayers. From such results, authors have argued for complete anion exclusion from interlayers, and thereby supported ideas of a multi-porous structure of compacted water saturated bentonite. It is, however, glaringly obvious that these simulations are not even close to being converged, and that they should not have been published in the first place. It is also clear that chloride does enter interlayers in properly conducted MD studies, in both tri- and bi-hydrated sodium montmorillonite.
Reasonably, it should only be a matter of time before researchers that support ideas of complete exclusion manage to perform MD simulations that better reflect anion equilibrium in montmorillonite. As such possible future simulations will confirm that anions have access to interlayers,1 I have in the back of my mind wondered about potential consequences. Will earlier publications be retracted? Will the entire “mainstream view” of the structure of compacted bentonite fall into oblivion? (I wish.) From this perspective I find it a bit amusing that no further MD simulation has been published to support complete anion exclusion for the last ten years (as far as I’m aware).
Hsi15 simulated bi-hydrated sodium montmorillonite interlayers in contact with a bulk compartment with two different NaCl concentrations (1.67 M and 0.55 M), and showed that these systems obey the rules for Donnan equilibrium, albeit with a substantial non-electrostatic contribution to the free energy (non-ideal conditions). Hsi22 continue this work by presenting a complementary simulation at a third NaCl concentration (1.0 M), and by performing corresponding simulations for CaCl2, at bulk concentrations 0.14 M, 0.28 M, 0.52 M, and 0.84 M (with all interlayer cations then being calcium, obviously). With chloride equilibrium simulations for several background concentrations for both Na- and Ca-montmorillonite, but for otherwise identical systems, Hsi22 are able to make thorough comparisons with Donnan equilibrium theory.
Chloride equilibrium — just as any other ion equilibrium — is conveniently expressed via the ratio \(\bar{\mathrm{c}} / c^\mathrm{ext}\), where \(\bar{\mathrm{c}}\) is the clay concentration and \(c^\mathrm{ext}\) is the corresponding bulk concentration. In a Donnan equilibrium context this concentration ratio may be identified with the ion equilibrium coefficient2 \begin{equation} \Xi_\mathrm{Cl} \equiv \frac{c^\mathrm{int}_ \mathrm{Cl}} {c^\mathrm{ext}_\mathrm{Cl}} \end{equation} where \(c^\mathrm{int}_\mathrm{Cl}\) is the interlayer concentration of chloride in a homogeneous bentonite domain in equilibrium with an external solution with chloride concentration \(c^\mathrm{ext}_\mathrm{Cl}\).
For a 1:1 system (e.g. NaCl in contact with Na-montmorillonite) a good approximation for \(\Xi_\mathrm{Cl}\) at low external concentration is3 \begin{equation} \Xi^{1:1}_\mathrm{Cl} \approx \Gamma^2 \frac{c^\mathrm{ext}_\mathrm{Cl}}{c_\mathrm{IL}} \tag{1} \end{equation} where \(c_\mathrm{IL}\) is the structural montmorillonite charge expressed as a monovalent interlayer concentration (in the model of Hsi22 and Hsi15, \(c_\mathrm{IL} = 4.23\) M) and \(\Gamma\) is a mean activity coefficient ratio for NaCl (more on that below).
While the ion equilibrium coefficient in eq. 1 depends linearly on the external concentration, the corresponding quantity for a 2:1 system (e.g. CaCl2 in contact with Ca-montmorillonite) depends on the square-root of the external concentration (note that the Cl concentration in a CaCl2 solution is twice that of CaCl2) \begin{equation} \Xi^{2:1}_\mathrm{Cl} \approx \Gamma^{3/2} \sqrt{\frac{c^\mathrm{ext}_\mathrm{Cl}}{c_\mathrm{IL}}} \tag{2} \end{equation} where \(\Gamma\) here is to be understood as a different mean salt activity coefficient ratio (for CaCl2).
The different dependencies on \(c^\mathrm{ext}_\mathrm{Cl}\) for Na- and Ca-systems, expressed in eqs. 1 and 2, are clearly reproduced in the MD results presented in Hsi22 as shown here
The dots show the chloride equilibrium coefficients as calculated in Hsi22 and Hsi15, primarily from evaluated potentials of mean force evaluated using the adaptive biasing force method. The corresponding curves in the above diagram are my attempt at fitting eqs. 1 and 2 to these MD results.
It should be noted that the linear and square-root dependencies of eqs. 1 and 2, respectively, presume that the activity coefficient ratios (\(\Gamma\)) are essentially independent of \(c_\mathrm{Cl}^\mathrm{ext}\). The successful fits of eqs. 1 and 2 thus demonstrate that this is the case for the MD equilibrium coefficients.4 Hsi22 make a deeper analysis and show that the specific values of the activity coefficient ratios correspond to differences in excess chemical potential for the salt of 1.35 kT and 1.25 kT, respectively, for the Na- and Ca-systems. Such values reflect a quite profound non-ideal behavior, which may be related to the details of the simulations (e.g. non-polarizable force fields) rather than corresponding to an actual excess barrier.
The main message in Hsi22 is nevertheless clear: Results from MD
simulations of chloride in Na- and Ca-montmorillonite are consistent
with Donnan equilibrium theory. This means, in particular
\(\Xi_\mathrm{Cl}\) is linear for NaCl and has a square-root dependence for CaCl2
For a given external chloride concentration and density, the amount chloride entering the interlayers is much larger in Ca-montmorillonite as compared to Na-montmorillonite
To be clear, the much larger amount of chloride predicted to be found
in Ca-montmorillonite has nothing to do with any notions of different
“anion-accessible” pore spaces, but is a direct consequence of
Donnan equilibrium. In these simulations, all chloride is located at
the exact same place within the clay, as shown here
This figure shows evaluated chloride density profiles in the direction perpendicular to the mineral layers in MD simulations of Na-montmorillonite (Hsi15) and Ca-montmorillonite (presented in the supporting information to Hsi22). While I have arbitrarily scaled the profiles along the y-axis in the above figure for visualization purposes, emphasis is here on the identical position within each interlayer. Note that the simulated system contains two separate interlayers, indicated by dotted vertical lines.5
One lesson from these results is that researchers who struggle with getting chloride to enter interlayers in their simulations could use CaCl2 rather than NaCl. At e.g. an external chloride concentration of \(\sim\)0.5 M, the amount chloride in the clay is about seven times larger in Ca- as compared with Na-montmorillonite, which substantially reduces the required convergence time for the simulation.
These results also highlight the urgent need for empirical data. As I
pleaded for when
concluding the assessment of chloride equilibrium concentrations in Na-bentonite, labs all over the place should routinely produce and
publish ion equilibrium measurements. It is certainly a failure of the
bentonite research field that no published empirical data exists that
can be used to compare with these theoretical results. Indeed, as far
as I’m aware, no published systematic empirical data exists at all,
for anion equilibrium concentrations in calcium dominated
bentonite.6
Note that the implication of the results discussed here is not simply
some noted interesting difference in chloride equilibrium in different
types of montmorillonite. Rather, as the results indicate that
montmorillonite interlayers play by the rules of ordinary Donnan
equilibrium, they are an additional blow to the entire contemporary
multi-porous model description of compacted water saturated bentonite.
Footnotes
[1] As I often nag about on this blog, it is quite silly to use complete anion exclusion as a starting point when studying compacted bentonite, and then trying to “confirm” such a notion with e.g. MD simulations. There is no rationale for this assumption in the first place; as we have discussed earlier, the idea seems to have originated from misunderstanding the Poisson-Boltzmann equation. Moreover, there is solid empirical evidence for salt entering interlayers, in particular from measured swelling pressure response.
[2] Hsiao and Hedström (2022) call this ratio a partition
coefficient, which complies with the scientific literature on
e.g. polymer membranes. As I discussed
here, I have chosen to stick with some of my own terminology. I
hope this does not cause unnecessary confusion.
[4] That the activity coefficient ratios do not depend strongly on external concentration in this concentration interval is also compatible with the mean salt approach that I have suggested to use for compacted bentonite. For the external solutions, mean salt activities varies quite little in this concentration range, and since the interlayer concentrations only vary with a few percent, it make sense to assume that the interlayer activity coefficients basically remain constant. Hsiao and Hedström (2022) actually note that the undulation pattern in the potential of mean force in the direction of the reaction coordinate is essentially independent of the external solution, and conclude that the interlayer environment is essentially independent of external conditions.
[5] The nearly
identical profiles within each interlayer is also a confirmation
that these simulations are properly converged.
[6] An indication that CaCl2 in Ca-montmorillonite behaves as discussed is found here.
The subsection we focus on here, “Adsorption processes in clays”,
contains very little descriptions of fundamental properties of
bentonite, and is instead almost exclusively devoted to detailed
discussions on various models. As an example, already in the
first paragraph the text digresses into dealing with the problem of
defining “surface species activity” in the “DDL”2 model…
TS15 discuss adsorption separately on “outer basal surfaces”, “interlayer basal surfaces”, and “edge surfaces”. Note that the distinction between “outer” and “interlayer” basal surfaces requires that we view the compacted bentonite as composed of stacks (referred to as “particles” in TS15). But this idea is just fantasy, as we have discussed in the previous part and in a separate blog post. Moreover, central to the description of adsorption processes in TS15 is the idea of a Stern layer. This concept was briefly introduced in the previous subsection (“Electrostatic properties, high surface area, and anion exclusion”)
The [electrical double layer] can be conceptually subdivided into a Stern layer containing inner- and outer-sphere surface complexes […] and a diffuse layer (DL) containing ions that interact with the surface through long-range electrostatics […].
The next time this concept is brought up is at the beginning of the
discussion on adsorption on “outer basal surfaces”
The high specific basal surface area and their electrostatic properties give rise to adsorption processes in the diffuse layer, but also in the Stern layer.
I have written a separate blog post arguing for that the idea of Stern layers on montmorillonite basal surfaces is unjustified. Note that the notion of Stern layers on montmorillonite basal surfaces in the contemporary bentonite literature de facto means that these surfaces are supposed to be full-fledged chemical systems. In particular, the basal surface is supposed to contain localized “sites” that interact generally with ions to form surface complexes and that can involve covalent bonding.
Note further that the Stern layer was originally introduced as a model (or a model component) that extends the Gouy-Chapman description of the electric double layer. TS15, on the other hand, use the term “Stern layer” to refer to an actual physical structural component. And just as in the case of several other “components” that has been introduced in the article (“particles”, “inter-particle water”, “free or bulk water”, “aggregates”…), the existence of a Stern layer is just declared rather than argued for. And just like with the other components, these are not universally adopted. I don’t think it is appropriate to include Stern layers in this way in a review article when established parts of the colloid science community refer to them as an “intellectual cul de sac”.
So in order to even begin to criticize what TS15 actually write about adsorption processes here, one has to accept both the flawed idea of stacks as fundamental structural units and the far from universally accepted idea of Stern layers on montmorillonite basal surfaces. I will therefore refrain from doing that, and simply proclaim that I don’t accept the premises. (I believe I will have reasons to return to the models presented here when reviewing later sections of TS15.)
Additional remarks
But I think it is worth reminding ourselves that at the end of the previous section (covered in part I) we were promised that this section should qualitatively link “fundamental properties of the clay minerals” to the diffusional behavior of compacted bentonite. A reader of TS15 will thus expect this section to contain, in particular, a reasonable description and discussion on how compacted montmorillonite works. Instead a very specific (and flawed) model is imposed on the reader: the first subsection (covered in part II), introduced the fictional stack concept, and gave a confused and irrelevant explanation of anion exclusion; the presently discussed subsection is centered around Stern layers.
If the authors truly did what they claimed, in this section they should have addressed the consequences of montmorillonite TOT-layers being charged — a universally accepted fact — without introducing further assumptions. This would naturally lead to a discussion on osmosis, swelling, swelling pressure and semi-permeable boundary conditions (all simple empirical facts). These topics, in turn, should lead to considerations of e.g. ionmobility and chemical interface equilibrium. Not a single one of these topics are, in any meaningful sense, actually addressed in this section.
Before ending this part of the review, I also would like to focus on what is being said bout “interlayers”. We should keep in mind that TS15 — together with a large part of the contemporary bentonite research community — assume “interlayers” to be something different than simply the space between adjacent basal surfaces: these are supposed to be internal to the fantasy construct of a stack. When discussing adsorption in these presumed compartments they write
The interlayer space can be seen as an extreme case where the
diffuse layer vanishes leaving only the Stern layer of the adjacent
basal surfaces.
Of everything I’ve read in the bentonite literature, this is the closest I’ve come to see some actual description of what the fundamental difference between an “outer basal surface” and an “interlayer” is supposed to be. But let’s think this through. TS15 have claimed that an electric double layer is composed of a Stern layer and a diffuse layer, and we have vaugley been told that ions in the Stern layer are immobile. The above quotation thus implicitly says that that “interlayer” ions are not mobile, and that diffuse layers are only supposed to exist on “outer basal surfaces” (which, remember, is a fantasy component). But — disregarding that the stack-internal “interlayer” also is a fantasy concept — it is an indisputable experimental fact that has been known for a longtime that interlayers provide the only relevant transport mechanism in compacted bentonite.
Thus, either TS15 here provide us with yet another incorrect description of the behavior of compacted bentonite (that “interlayer” ions are immobile) or they are claiming, somewhat contradictorily, that Stern layer ions are mobile after all. But if Stern layer ions diffuse, such a structural component could reasonably not have been singled out in the first place! (The diffuse layer is supposed to have “vanished”.) As with many other issues in TS15, this question is left vague and unanswered.3 The continuation of the text does not make things clearer
For this reason, the interlayer space is often considered to be
completely free of anions (Tournassat and Appelo 2011), although
this hypothesis is still controversial (Rotenberg et al. 2007c;
Birgersson and Karnland 2009).
An interlayer completely devoid of anions certainly play by other rules than an “ordinary” electric double layer. Does this mean that TS15 assume “interlayer” ions to be immobile?4 Anyway, it is an indisputableexperimental fact that anions occupy interlayers, and I find it quite bizarre to find myself referenced in connection with the “controversial hypothesis”. The idea of compartments completely devoid of anions is widespread in the contemporary bentonite research community, but no one has ever suggested a mechanism for how such an exclusion is supposed to work; here, it apparently should be related to “Stern layers” in some (unexplained) manner. At the same time, the simplest application of Donnan equilibrium principally explains e.g. the behavior of the steady-state flux in anion tracer through-diffusion tests.
The agreement between [Poisson-Boltzmann] calculations and MD
simulation predictions was somewhat worse in the case of the
\(\mathrm{Cl^-}\) concentration profiles than in the case of the
\(\mathrm{Na^+}\) profiles (Figure 3), perhaps reflecting the poorer
statistics for interlayer Cl concentrations or the influence of
short-range ion-ion interactions (and possibly ion- water
interactions, as noted above) that are not accounted for in the
[Poisson-Boltzmann] equation. Nevertheless, reasonable quantitative
agreement was found (Table 2).
Here they acknowledge not only that anions do occupy interlayers, but also that the interlayer plays by the same rules as the “ordinary” electric double layer (“Poisson-Boltzmann calculations”). What happened to the “vanishing” diffuse layer, and to “considering” the interlayer to be “completely free of anions”? I find it quite outrageous that they fail to acknowledge these blatantly mixed messages with so much as a single word.
Update (251106): Part IV of this review is found here.
Footnotes
[1] As I have commented in the
earlier parts: TS15 are fond of using the very general terms
“clays” and “clay minerals”, while it is clear that the
publication mainly focus on systems with substantial ion exchange
capacity and swelling properties. Here we will continue to use the
term “bentonite” for these systems, and ignore the frequent
references in TS15 to more general terms.
[2] For some
reason, “DDL” is short for (the very generically sounding) “double
layer model”. Why not “DLM”?
[3] Spoiler: in later sections describing models, TS15 allow for the possibility of transport in “interlayers”.
[4] Questions like these can often not be answered because so many statements in TS15 are vague and ambiguous. In this discussion we have to refer to statements such as (my emphasis)
“The EDL can be subdivided into a Stern layer […] and a diffuse layer […].”
“The interlayer can be seen as an extreme case where the diffuse layer vanishes […]”
“The interlayer space is often considered to be completely free of anions […]”
I get annoyed by too much of such language in scientific
publications.
I argue that the only significant pore type in water saturated compacted bentonite is interlayers, by which I mean pores where the exchangeable cations reside (together with any other dissolved species). From this perspective it naturally follows that a homogeneous view is a suitable starting point for modeling compacted bentonite. I have presented, used, and discussed the homogeneous mixture model in many places on the blog, the main sources being
For reasons I can’t get my head around, a homogeneous view of
compacted bentonite is not the mainstream in contemporary
bentonite research. Instead we are stuck with
“the mainstream view”, which postulates several distinctly
different pore structures within the bentonite; in particular, the
mainstream view uses a bulk water phase as a starting point and also
distinguishes between “outer” and “inner” basal surfaces. Electric
double layers are assumed to only exist on “outer” surfaces, while
the function of the “inner” basal surfaces is mostly shrouded in
mystery.
On the blog I have also presented plenty of experimental support for a
homogeneous view. A main argument is that the conditions for swelling
pressure — the most profound feature of bentonite in equilibrium with an external solution — are essentially fulfilled automatically in the
homogeneous mixture model. The mainstream view, in contrast, requires
handling of the seemingly contradictory situation of having swelling
pressure while the water chemical potential is supposedly restored
without pressurization. Proponents of the mainstream view often deal
with this by simply ignoring swelling phenomena altogether.
I have also on the blog dissected several studies that argue for a
non-homogeneous view, but that actually provide evidence for the
opposite when examined more carefully. Consider in particular:
By systematically varying background concentration, material, and diffusing tracer, Glaus et al. (2007) clearly demonstrate, not only that the exchangeable cations are mobile, but that they dominate the flux in through-diffusion tests in highly compacted montmorillonite. While this certainly is an argument for that compacted bentonite is homogeneously structured, Glaus et al. (2007) still analyze their results from the perspective of the mainstream view, and do not — in my view — fully conclude what their results imply.
In particular they postulate the presence of an interlayer domain and a “free pore water” domain, and write for the “total” flux1 (their eq. 3)
where \(J_\mathrm{il}\) is a presumed diffusive flux in the interlayer domain and \(J_\mathrm{pw}\) is the presumed diffusive flux in the “free pore water” domain.
Their subsequent analysis shows that the measured flux in montmorillonite scales as
where \(C_\mathrm{bkg.}\) is the concentration of the background electrolyte (NaClO4), and \(Z\) is the charge number of the diffusing tracer (\(Z = 1\) for sodium and \(Z=2\) for strontium). Moreover, by considering ion exchange equilibrium, Glaus et al. (2007) show that also \(J_\mathrm{il}\) is expected to scale according to eq. 2. As they also confirm that this scaling behavior is not observed in systems without interlayer pores (kaolinite), they could have confidently concluded that their results imply that interlayers are the only significant pore structure in montmorillonite at these densities (as the title suggests).
Unfortunately, the discussion part of the article is considerably more tentative, focusing mainly on “interpretations” of the resulting flux
The present work shows that the interpretation of cation diffusion experiments in highly compacted swelling clays in terms of the concentration gradient in the aqueous phase may result in a nonsensical dependence of the effective diffusion coefficients on the salt concentration in the external aqueous phase. An alternative interpretation using an effective diffusion coefficient in the interlayer water (\(D_\mathrm{il}\)), being independent of the external salt concentration, with a corresponding concentration gradient in the interlayer water is more consistent with the experimental observations.
and the article ends on a quite apologetic note
The proposed interpretation should in turn not be blindly applied to
other experimental conditions. Diffusion of cations via the free
pore water may become increasingly important in swelling clays with
lower degrees of compaction or in clays in which the interlayer gel
pores are not that adjacent as they are in compacted
montmorillonite. In such cases, the assumption of
\(J_\mathrm{tot} \cong J_\mathrm{il}\) may no longer hold, and a
double-porous diffusion model would have to be applied in such
cases. The present concept may also reach its limits when dealing
with cations that rather sorb by surface complexation than by ion
exchange. Further work is therefore planned to extend the
investigations to such systems.
Given that the mainstream view to this day continues to be the default approach, one may think that this “further work” did show some convincing evidence for e.g. “diffusion of cations via the free pore water” at lower density. But what has actually been shown is that the “assumption of \(J_\mathrm{tot} \cong J_\mathrm{il}\)“ continues to be true for lower density!
Before we look at the additional results, we summarize the findings of
Glaus et al. (2007).
Findings in Glaus et al. (2007)
In the following we will consider the so-called “effective diffusion
coefficient”, here strictly defined as the experimental parameter
where \(j_\mathrm{ss}\) denotes the steady-state flux when an external
tracer concentration difference \(\Delta c^\mathrm{ext}\) is maintained
across a bentonite sample of length \(L\). We have
discussed through-diffusion and
the role of \(D_e\) in
many places on the blog, but in the present discussion we simply
view \(D_e\) as a normalized version the steady-state flux.
Note that we are required to compare diffusive fluxes in
different montmorillonite samples (an alternative test protocol
is suggested below). \(D_e\) varies both due to varying background
concentration (which is our object of study) and due to the variation
of different samples. It is thus crucial to minimize the latter type
of variation. This should be done (I suppose) by employing as
identical preparation protocols as possible. We will get back to this
complication of sorting out signal from noise as we comment the
results.
Glaus et al. (2007) present their results in diagrams where the logarithm of the evaluated quantities (diffusion parameters) is plotted against background concentration. This is of course convenient, as e.g. \(D_e\) can be expected to vary by two orders of magnitude as the background concentration is varied between 0.01 M and 1.0 M. But to remind ourselves what the actual dependency looks like between the normalized steady-state flux and background concentration, I will here insist on plotting the results in lin-lin diagrams.
The results for sodium in Glaus et al. (2007) plotted in lin-lin
diagrams, look like this (the data is the same in these three
diagrams)
We see that the data complies with the scaling law (eq. 2) and is quite well constrained (click on pictures to enlarge). \(D_e\) is evaluated in two ways in Glaus et al. (2007): by examining at the breakthrough curve, and by examining the internal tracer profile at test termination. These methods of evaluation give more or less identical results, with the exception of the test performed at 0.01 M background concentration. In this low concentration limit, the confining filters increasingly restrict the flux, making it difficult to extract actual clay transport parameters. We have discussed this issue (and this particular study) at length in a previous blog post.
Even with the problem of accurately measuring \(D_e\) at the lowest background concentration, the results clearly demonstrate the behavior of a homogeneous system (eq. 2): e.g., \(D_e\) undoubtedly increases by a factor of approximately 10 when the background concentration is lowered from 1.0 M to 0.1 M.
The data for strontium in Glaus et al. (2007) only covers the
background concentration interval 0.5 M — 1.0 M, and is consequently
less constrained, as seen here
This data also has the peculiarity that the diffusivity of samples of length 5.4 mm is almost twice as large as for samples of length 10.4 mm. This clearly demonstrates how sample preparation becomes crucial when conducting these types of tests. In the plots above, I have allowed myself to treat samples of different length separately (Glaus et al. (2007) use average values). It is clear from the data, that also strontium is compatible with the scaling law of eq. 2. In particular, it can be distinguished that sodium and strontium have different dependencies.
The take away message from these results is clear: montmorillonite at this density (1950 kg/m3) behave as a homogeneous system and shows no indication of containing additional pore structures.
Glaus et al. (2013) and NTB-17-12
After the publication of Glaus et al. (2007), corresponding results for lower densities has been presented. Glaus et al. (2013) — which is mostly recognized for demonstrating the seeming “uphill” diffusion effect — also contains measured \(D_e\) of sodium as a function of background concentration in conventional through-diffusion tests, both for density 1600 kg/m3 and 1300 kg/m3. These results are also published in more detail,2 together with new strontium results, in the NAGRA technical report NTB-17-12. We therefore look at these two publications together.
The additional data for sodium is here compared with the results from
Glaus et al. (2007)
For some of the additional tests, both through- and out-diffusion were performed. These points are labelled “TD” and “OD”, respectively, in the diagrams. We see that even for density as low as 1300 kg/m3, the data complies with the behavior of a homogeneous system (eq. 2) and is quite well constrained; in particular, there is nothing in the data for 1300 kg/m3 that suggests that these systems behave principally different than the 1950 kg/m3 samples.
For the system at 1300 kg/m3 and background concentration 0.1 M, two different values of \(D_e\) are presented in NTB-17-12. Only the lower of these values (\(7.0\cdot 10^{-10}\) m2/s) was published in Glaus et al. (2013), but NTB-17-12 presents a continued analysis that includes filter resistance, giving the value of \(D_e\) presented in the diagram. I think this is quite interesting, as the tests made at 0.1 M used “flushed” filters in order to minimize filter resistance. Apparently, filter resistance is still influential and it is not that easy to “design away” this problem.
NTB-17-12 also presents measured values of \(D_e\) for strontium under similar conditions (1300 — 1900 kg/m3, 0.1 — 1.0 M NaClO4 background), and are here compared with the earlier results
Although it naturally contains some scatter, we note that the additional data for ~1900 kg/m3 strengthens the earlier conclusion that also strontium scales in accordance with eq. 2. And just as for sodium, we see that the behavior does not qualitatively change, even for densities as low as 1300 kg/m3.
In the above diagrams are plotted single values for \(D_e\) for strontium at the lowest background concentration (0.1 M). It should be noted that these are burdened with large uncertainties as the transport restriction of the confining filters is severe; in NTB-17-12 are presented a whole set of simulations of the underlying flux evolution and concentration profiles with variations of the filter transport parameters. It is thus very clear that the problem of eliminating transport restrictions at the sample interfaces are not easy to completely eliminate. This is not surprising, as the theory suggests that \(D_e\) increases without limit with decreasing background concentration. Note that this behavior is strongly enhanced for divalent strontium; the measured values are many times larger than the corresponding diffusivity in bulk water (\(0.79\cdot 10^{-9}\) m2/s).
Even if the value of \(D_e\) is quite uncertain at the lowest background concentration, the mere observation that filter diffusivity strongly influence the process is, in a sense, itself a confirmation that the system still is governed by the behavior of interlayers.
The picture is quite clear from these findings: the combined results of Glaus et al. (2007), Glaus et al. (2013) and NTB-17-12 validates a homogeneous view of compacted bentonite, at essentially any relevant density!
The curious case of Bestel et al. (2018)
Bestel et al. (2018) further examine how \(D_e\) for sodium varies with background concentration. This publication shares some of the same authors with the previous studies, and presents additional measurements of \(D_e\) for sodium in essentially identical systems (similar preparation protocols, “Milos” montmorillonite, NaClO4 background electrolyte, flushed filters). Given the substantial evidence for homogeneous behavior collected in the publications discussed above, I find the conclusions of Bestel et al. (2018) rather odd.
Bestel et al. (2018) perform subsequent measurements of the steady-state flux in the same samples at different temperatures. The dependency of \(D_e\) on background concentration, however, looks essentially the same for each temperature, and — just as Bestel et al. (2018) — we here focus mainly on the results for 25 \(^\circ\mathrm{C}\). This data looks like this3
In their analysis, Bestel et al. (2018) include the results from Glaus et al. (2007) and Glaus et al. (2013), but treat them separately. They consequently conclude implicitly that, although the earlier studies found that \(D_e\) depends on background concentration in accordance with eq. 2, the new results show a different behavior. Specifically, they conclude that \(D_e\) scale with background concentration as \(C_\mathrm{bkg}^{-0.52}\) for density 1300 kg/m3 and as \(C_\mathrm{bkg}^{-0.76}\) for density 1600 kg/m3. Bestel et al. (2018) write
The results obtained in the present work for a broad variety of bulk
dry densities of Na-montmorillonite and concentrations of the
background electrolyte, give clear evidence that the equilibrium
distribution of cations between the clay phase and the external
aqueous phase is the main parameter influencing the observed overall
diffusive fluxes of cations. Whether the observed overall diffusive
fluxes are described by a physical subdivision of the pore space
into domains containing different species (e.g. the model proposed
in Appelo and Wersin (2007) or Bourg et al. (2007)), or whether
they are the result of the concentration gradients of such species
in a single type of pore (e.g. the model proposed by Birgersson and
Karnland (2009)), cannot be decided unambiguously from the available
data — notably because of the wide similarity of the model
predictions and because of some internal inconsistencies in the
experimental data. Both types of models would require some
adjustments in order to fully match the data. The diffusion data of
\(^{22}\mathrm{Na}^+\) can equally be described by a surface diffusion
model with a reduced, but non-zero mobility of sorbed cations,
similar to the median value determined in Gimmi and Kosakowski
(2011).
I think this is a problematic way of arguing and presenting data.
The data obviously has scatter
To begin with, why are the results from this study and the ones from Glaus et al. (2007) and Glaus et al. (2013) treated separately? When treated separately — according to Bestel et al. (2018) — these results are vaguely supposed to be incompatible: the dependence of \(D_e\) either comply with eq. 2 or it does not. I think that the appropriate thing to do is to discuss possible causes for why the new results supposedly differ from the earlier ones. As we have made clear above, all factors that determine \(D_e\) are not fully controlled in tests like these (e.g, what causes the difference in diffusivity for strontium in 5.4 mm and 10.4 mm samples, respectively, in Glaus et al. (2007)?). We have also seen that it is difficult to make accurate measurements at low enough background concentration, even with flushed filters.
Look e.g. at the specific values of \(D_e\) at background concentrations 1.0 and 0.1 M, respectively, in NTB-17-12 and Bestel et al. (2018) (unit is m2/s).
Under ideal conditions, these values would not differ for the same conditions in the two studies. The scatter of these values is moreover quite random, e.g. one study do not have values that are systematically larger than in the other. In Bestel et al. (2018) we also see that the mere disturbance of a sample in form of a temperature pulse may alter the diffusivity significantly (temperature is first increased in steps from 25 \(^\circ\mathrm{C}\) to 80 \(^\circ\mathrm{C}\), then decreased in steps to 0 \(^\circ\mathrm{C}\), and finally increased again to 25 \(^\circ\mathrm{C}\)). In e.g. one sample of density 1600 kg/m3 and background concentration 0.1 M is reported \(D_e = 3.4\cdot 10^{-10}\) m2/s at 25 \(^\circ\mathrm{C}\) before the conducted temperature changes, and \(2.3\cdot 10^{-10}\) m2/s after. One should also consider that the samples are not prepared equally, as they are saturated directly with the corresponding background solution. (This is also true for the previous studies.) Could this cause differences in diffusivity?
Bestel et al. (2018) should thus either argue for why the new results
are more accurate (or why the results of Glaus et al. (2007, 2013)
are less accurate) or treat the data from all studies in accumulation
and admit substantial experimental uncertainty. My impression is that
Bestel et al. (2018) make a little of both.
The data still complies with a homogeneous view
Looking at the aggregated sodium data, a somewhat different picture
emerges
Here is also included a model labelled “Full Donnan”, which takes into account the excess salt that is expected to enter the interlayers. For all other samples we have discussed, this contribution is only minor and can be neglected, and this assumption underlies eq. 2. For the sample of density 1300 kg/m3 with background concentration 5.0 M, however, the excess salt is not negligible and must be included in the analysis of the behavior of a homogeneous system (the deviation from eq. 2 is seen to become significant around 1.0 M background concentration). Bestel et al. (2018) actually present a full Donnan calculation for the excess salt, but, for unknown reasons, do not compare it directly with the experimental results (it is plotted in a separate diagram next to the data).
For 1300 kg/m3, I would claim that the “Full Donnan” model fits better to the accumulated data than the scaling law suggested in Bestel et al. (2018) (exponent \(-0.52\)). For 1600 kg/m3, the suggested scaling law (exponent \(-0.76\)) indeed fits better to the data than eq. 2, but the data is not that well constrained. To use this singular result to argue for a non-homogeneous bentonite structure basically boils down to claiming that the values measured at 0.1 M — a concentration range that is documented to be difficult to measure accurately — could not possibly be underestimated by, say, 50% (while also ignoring all other results).
If we also consider the results for strontium presented in NTB-17-12, I mean that the only reasonable conclusion that Bestel et al. (2018) can draw is that the results comply with a homogeneous bentonite structure.
Additional model components should not be motivated
solely by the ability of a model to be fitted to some arbitrary
data
A major motivation for measuring how \(D_e\) depends on background concentration at lower densities, according to Glaus (2007), is that “the assumption of \(J_\mathrm{tot} \cong J_\mathrm{il}\) may no longer hold”. What (I mean) has been demonstrated in the subsequent studies is that this assumption actually does hold. In particular, from the aggregated data it is not possible to claim that the behavior of \(D_e\) is qualitatively different at 1950 kg/m3 and 1300 kg/m3. Thus, there is no valid justification for introducing more complex model components. Moreover, introducing e.g. a bulk water phase causes fundamental conflicts with the description of other well-established properties of these systems, particularly swelling pressure. Adding such components merely to improve agreement with a specific dataset, while ignoring their broader implications, undermines the model’s overall coherence and validity. The data cannot“equally be described by a surface diffusion model”.
What does some alternative model actually predict?
Eq. 2 (or a full Donnan calculation) is a clean statement of the expected behavior of a homogeneous system (based on how interlayers function). If actual deviations from this behavior could be established we may conclude that a homogeneous description is not sufficient. However, any arbitrary deviation from eq. 2 does not automatically validate any specific alternative model. Validating a model requires that we can experimentally reproduce some of its non-trivial predictions. Bestel et al. (2018) don’t discuss what the exponents \(-0.52\) and \(-0.76\) are suppose to represent.
Note also that the arbitrary exponent \(-0.52\) is inferred by fitting to the data at 5.0 M background concentration. But we saw above that a full Donnan calculation within the homogeneous view actually explains the behavior in this concentration limit (Bestel et al. (2018) show this!). We have thus every reason to believe that the exponent \(-0.52\) is just spurious and do not represent some actual physical mechanisms.
A suggestion for how to preferably conduct these types of tests
The discussed studies are enough to convince me that cation tracer
diffusion behave in accordance with a homogeneous bentonite view at
any relevant density.
It is however also clear that the full variation of \(D_e\) in these
tests is caused by more factors than just background concentration and
density. To eliminate as much as possible of this scatter — and thus
to more accurately determine the dependence of background
concentration on \(D_e\) — I suggest the following test protocol.
Measure tracer flux at several background concentrations in the same sample.
This would eliminate both the unavoidable (small) variation in density between different samples as well as several unknown factors that determine the exact value of diffusivity (these may e.g. be related to variation in material or equipment and to sample handling)
Prepare samples by saturating them all with the same low concentration solution (e.g. 0.05 M).
To me it seems reasonable that the way samples saturate may influence the resulting detailed structure and thus the diffusivity. Saturating all samples in the same manner with the same solution will minimize variations from such effects.
Keep temperature constant.
I don’t think this is a crucial factor, but we see in Bestel et al. (2018) that larger temperature pulses may significantly alter the diffusivity.
Increase background concentration in steps and record the steady-state flux at each concentration.
I think a good range may be between 0.2 M and 1.0 M. For a homogeneous system, this corresponds to a variation in \(D_e\) by a factor 5 for monovalent and 25 for divalent cations.5 At the same time, the problem of filter transport resistance can hopefully be kept under control.
Decrease the background concentration (perhaps in steps) back to the first concentration where steady-state flux was measured.
Measure steady-state flux again and assert that no significant change in \(D_e\) has occurred as a consequence of the disturbance introduced by the background concentration pulse.
Final thoughts
The only reasonable conclusion to draw from the studies we have looked
at is that the behavior of cation tracer diffusion indicates a
relatively homogeneous structure, dominated by interlayers, in any
relevant bentonite system. Despite this, the contemporary scientific
bentonite literature is crammed with non-homogeneous descriptions of
compacted bentonite, centered around a bulk water phase (the
“mainstream view”). As we have seen here, this can even be the case
for studies that provide evidence for homogeneity.
What I find most frustrating is that interlayer effects often are viewed as some additional feature to be handled in specific cases. In reality, virtually all experimental findings (diffusion, swelling pressure, temperature response, Donnan effects, fluid flow, hyperfiltration, …) indicate that the behavior of compacted bentonite is fully governed by interlayers. The question is not if a presumed bulk water phase may dominate under certain conditions, but if such a phase is at all relevant. I want to emphasize this point: up until this day, no convincing evidence has ever been presented that compacted bentonite contains significant amounts of bulk water.
Even if the structure becomes more complex at lower densities, a
homogeneous model centered around interlayers guarantees to cover at
least some aspects of the system. On the contrary — if the goal is
process understanding — most experimental evidence rules out
bentonite models that assume a bulk water phase.
[2] As
far as I can see, these tests were done in duplicates for Na
diffusion with background concentrations 0.5 M and 1.0 M, and the
the numbers reported in Glaus et al. (2013) are averages.
[3] Bestel et
al. (2018) use a normalization scheme in their analysis that
involves corresponding measured water diffusivities and parameters
from “Archie’s law” (note, it is the
quotation marks version of the law). I think this handling makes
the presented results less transparent, and here we use the actual
reported values of \(D_e\).
[4] These are only values from the first phase at 25 \(^\circ\mathrm{C}\).
[5] I assume that measurements are being made in pure Na-montmorillonite.
This is the second part of the review of “Ionic Transport in Nano-Porous Clays with Consideration of Electrostatic Effects” (Tournassat and Steefel, 2015) (referred to as TS15 in the following). For background and context please check the first part. That part covered the introduction and the section “Classical Fickan Diffusion Theory”. The next section is titled “Clay mineral surfaces and related properties”, and is further partitioned into two subsections. Here we exclusively deal with the first one of these subsections: “Electrostatic properties, high surface area, and anion exclusion”. It only covers three and a half journal pages, but since the article here goes completely off the rails, there is much to comment on.
“Electrostatic properties, high surface area, and anion
exclusion”
As stated in the first part, I find it remarkable that the authors use
general terms such as “clay minerals” when the actual subject matter
is specifically systems with a significant cation exchange capacity,
and montmorillonite in particular. I will continue to refer to these
systems as “bentonite” in the following, disregarding the constant
references to “clay minerals” in TS15.
Stacks
After having established that montmorillonite and illite have
structural negative charge, it begins:
Clay mineral particles are made of layer stacks and the space
between two adjacent layers is named the interlayer space (Fig. 5).
This is the first mention of clay “particles” in the article, and they are introduced as if this is a most well-established concept in bentonite science (incredibly, it is also the first occurrence of the term “interlayer”). We will refer to “clay mineral particle” constructs as “stacks” in the following. I have written a detailed post on why stacks make little sense, where I demonstrate their geometrical impossibility and show that most references given to support the concept are studies on suspensions that often imply that montmorillonite do not form stacks. Sure enough, this is also the case in TS15
The number of layers per montmorillonite particle depends on the
water chemical potential and on the nature and external
concentration of the layer charge compensating cation (Banin and
Lahav 1968; Shainberg and Otoh 1968; Schramm and Kwak 1982a;
Saiyouri et al. 2000)
Banin and Lahav (1968), Shainberg and Otoh (1968), and Schramm and Kwak (1982) all report studies on montmorillonite suspensions. The abstract of Shainberg and Otoh (1968) even states “The breakdown of the tactoids occurred when the equivalent fraction of Na increased from 0.2 to 0.5. Montmorillonite clay saturated with 50% calcium (and less) exists as single platelets.”, and the abstract of Schramm and Kwak (1982) states “Upon exchange of Ca-counterions for Li-, Na-, or K-counterions, a sharp initial decrease in tactoid size was observed over approximately the first 30% of cation exchange.”. These are just different ways of saying that sodium dominated montmorillonite is sol forming.
I want to stress the absurdity of the description given in TS15. A pure fantasy is stated about how compacted bentonite is structured. As “support” for the claim are given references to studies on “dilute suspensions”. It should be clear that the way TOT-layers interact in such suspensions essentially says nothing about how they are organized at high density. But even if we pretend that these results are applicable, the given references say that most of the relevant systems (montmorillonite with about 30% sodium or more) do not form stacks.
Disregarding the references, note also how bizarre the above statement is that the number of layers in a “particle” depends on “the water chemical potential and on the nature and external concentration of the layer charge compensating cation”: stacks are supposed to be fundamental structural units, yet the number of layers in a stack is supposed to depend on the entire water chemistry?! (It makes sense, of course, for stacks in actual suspensions.) Also, for montmorillonite an actual number of layers is nowhere stated in TS15.
TS15 further complicate things by lumping together montmorillonite and
illite. In contrast to Na-montmorillonite, illite has by definition a
mechanism for keeping adjacent TOT-layers together: its layer charge
density is higher and compensated by potassium, which doesn’t hydrate
that well, leading to collapsed interlayers. As far as I understand,
one characterizing feature of illite is that the collapsed interlayers
are manifested as a
“10-angstrom peak” in X-ray diffraction measurements.
To treat montmorillonite and illite on equal footing (in a laid-back single sentence) again shows how nonsensical this description is. Stacking in montmorillonite suspensions occurs as a consequence of an increased ion-ion correlation effect when the fraction of e.g. calcium becomes large (> 70-80%). This process requires the ions to be diffusive and is distinctly different from the interlayer collapse in illite.
I actually have a hard time understanding what exactly is meant by the
term “illite” here. In clay science it is clear that what is
referred to by this name are systems that may have a quite
considerable cation exchange
capacity.1 Reasonably, such systems contain other types of cations
besides potassium2 (as they
are exchangeable), and must contain compartments where such ions can
diffuse (as they are exchangeable). To increase the complexity, there
are also “illite-smectite interstratified clay minerals”, which typically
are in “smectite-to-illite” transitional states. For these, it seems
reasonable to assume that the remaining smectite layers provide both
diffusable interlayer pores and the cation exchange capacity. I don’t
know if such “smectite layers” provides the cation exchange capacity
in general in systems that researchers call illite. Neither do I
understand how researchers can accept and use this, in my view, vague
definition of “illite”. Anyway, it is the task of TS15 to sort out
what they mean by the term. This is not done, and instead we get the
following sentence
Illite particles typically consist of 5 to 20 stacked TOT layers
(Sayed Hassan et al. 2006).
This study (Sayed Hassan et al., 2006) concerns one particular material (illite from “the Le Puy ore body”) that has been heavily processed as part of the study.3 I mean that such a specific study cannot be used as a single reference for the general nature of “illite particles”. Moreover, the stated stack size (5 — 20 layers) is nowhere stated in Sayed Hassan et al. (2006)!4
In their laid-back sentence, TS15 also implicitly define “interlayer space” as being internal to stacks. I criticized this way of redefining already established terms in the stack blog post, and TS15 serves as a good illustration of the problem: are we not supposed to be able to use the term “interlayer” without accepting the fantasy concept of stacks? To be clear, “interlayer spaces” in the context of montmorillonite simply means, and must continue to mean, spaces between adjacent TOT basal surfaces. It drives me half mad that the “stack-internal” definition is so common in contemporary bentonite scientific literature that this point seems almost impossible to communicate.
The provided illustration (“Fig 5”) explicitly shows how TS15 differ
between “interlayers” that are assumed internal, and “outer basal
surfaces” that are assumed external to the stack.
This illustration misrepresents the actual result of assembling a set of TOT-layers, just like any other “stack” picture found in the literature. The figure shows five identical TOT-layers that can be estimated to be smaller than 20 nm in lateral extension (while the text “conveniently” states that they should be 50 — 200 nm). Compared with “realistic” stacks, formed by randomly drawing TOT-layer sizes from an actual distribution, the depicted stack in TS15 looks like this5 (see here for details)
Besides the fact that “realistic” stack units cannot be used to form the structure of compacted bentonite, it should also be clear from this picture that “outer basal surfaces” and “interlayers” (in the sense of being internal to the stack) are not well defined. Note further that in actual compacted systems (above 1.2 g/cm3, say) such “realistic” stacks would be pushed together, something like this
In this picture, why should e.g. the interface between the green and the red stack be defined as an interface between two “outer surfaces” rather than an interlayer? Also, is this interface supposed to change nature and become an “interlayer”, as the water chemical potential or the external ion content changes? Like all other proponents of stack descriptions that I have encountered, TS15 do not in any way explain how “interlayers” and “outer surfaces” are supposed to function fundamentally differently. Similarly, they do not describe how the number of layers in a stack depends on water chemistry, nor do they provide a mechanism for why (sodium dominated) montmorillonite stacks of are supposed to keep together.
I want to emphasize that I do not favor any construction with
“realistic” stacks, but only use them to illustrate the absurd
consequences of taking a stack description seriously, and to
demonstrate that all such descriptions in the bentonite
literature are essentially pure fantasies, including the one given in
TS15. I’m also quite baffled as to why TS15 (and others)
provide such completely nonsensical descriptions, and how these can
end up in review articles. I believe a hint is given in this
formulation
[T]he number of stacked TOT layers in
montmorillonite particles dictates the distribution of water in two
distinct types of porosity: the interlayer porosity […]
and the inter-particle porosity.
The only way I can make sense of this whole description is as an
embarrassing attempt to motivate the introduction of models with
several “distinct types of porosity”: the outcome is simply a
macroscopic multi-porosity model (which will also be evident in later
sections).
I’ve written a detailed blog post on why multi-porosity models cannot be taken seriously. There I point out that basically all authors promoting multi-porosity for some reason attempt to dress it up in terms of microscopic concepts, while the models obviously are macroscopic. Moreover, no one has ever suggested a mechanism for how equilibrium is supposed to be maintained between the different types of “porosities”.
Anion exclusion
After hallucinating about the structure of compacted bentonite, TS15 change gear and begin an “explanation” of anion exclusion. Let’s go through the description in detail.
The negative charge of the clay layers is responsible for the
presence of a negative electrostatic potential field at the clay
mineral basal surface–water interface.
I cannot really make sense of the term “negative electrostatic
potential field”, although I think I understand what the authors are
trying to say here. What is true is that the electrostatic potential
near a montmorillonite basal surface is lowered compared to a
point farther away. But whether or not the value of the potential is
negative is irrelevant, as we are free to choose the reference
level. If the zero level is chosen at a point very far from the
surface, which often is done, it is true that the potential is
negative at the surface. But the key principle is that the potential
decreases towards the surface.6
A varying
electrostatic potential signifies an electric field, which in this
case is directed towards the surface (\(E = -d\phi/dx\)).
Furthermore, the electric field is not present merely because
of the presence of negative charge, but because this charge is
constrained to be positioned in the atomic structure of the
clay. Remember that the structural clay charge is compensated by
counter-ions, and that the system as a whole is charge neutral. The
reason for the presence of an electric field near the surface is due
to charge separation. And the reason for the potential
decreasing (i.e. the electric field pointing towards the surface) is
because it is the negative charge that is unable to be completely
freely distributed.7
The concentrations of ions in the vicinity of basal planar surfaces
of clay minerals depend on the distance from the surface
considered. In a region known as the electrical double layer (EDL),
concentrations of cations increase with proximity to the surface,
while concentrations of anions decrease.
Having established that the electrostatic potential varies in
the vicinity of the surface, it follows trivially that the ion
concentrations also vary. I also find it peculiar to label the regions
where the concentrations varies as the EDL. An electric double layer
is a structure that includes both the surface charge and the
counter-ions (hence the word “double”). What is described here
should preferably be called a diffuse layer. Note, moreover, that the
way an electric double layer here is introduced implies that TS15
consider a single interface, i.e. some variant of the Gouy-Chapman
model (this becomes clear below). But this model is not applicable to
compacted bentonite.
At infinite distance from the surface, the solution is neutral and
is commonly described as bulk or free solution (or water).
Here I think it becomes obvious that the authors try to motivate the presence of bulk water within the clay structure. As described in the blog post on “Anion accessible porosity”, it is only reasonable to assume that diffuse layers merge with a bulk solution in systems that are very sparse — i.e. in suspensions.8 This is how e.g. Schofield (1947) utilized the Gouy-Chapman model to estimate surface area. But how is the solution next to a basal surface in compacted bentonite supposed to merge with a bulk solution? Even if we use the authors’ own fantasy stack constructs, the typical structure of compacted bentonite must be envisioned something like this (I have color coded different stacks to be able to understand where they begin and end).
The regions where basal surfaces of different stacks face each other
(labelled A) are way too small in order to merge with a bulk solution
(and, as asked earlier, how are these regions even different from
“interlayers”?). Furthermore, regions adjacent to external
edge-surfaces of these imaginary stack units (B, C) are not at all
considered by applying a Gouy-Chapman model. The only way to make
“sense” out of the present description is to imagine larger voids in
the clay structure, something like this
But even if such voids would exist (in equilibrated water-saturated bentonite under reasonable conditions, they do not) they would only constitute an exotic exception to the typical pore structure. By focusing on this type of possible “anion” exclusion, TS15 completely miss the point.
This spatial distribution of anions and cations gives rise to the
anion exclusion process that is observed in diffusion experiments.
Now I’m lost. I don’t understand how ion distributions are supposed to cause a process. I think the authors here allude to Schofield’s approach to estimating surface area in montmorillonite suspensions. As discussed in detail in the blog post on anion-accessible porosity, if the suspension is so dilute that we can consider each clay layer independently, and if we equilibrate it with an external solution, we can measure its salt content, and use the Gouy-Chapman model to e.g. estimate surface area from the amount of excluded salt (as compared with the external solution).
But, as also discussed in the blog post on salt exclusion, the “Schofield type” of exclusion is not what we expect to be dominating in a dense system. Rather, in denser systems (and in Donnan systems generally — no surfaces need to be involved), salt exclusion occurs mainly because of charge separation at interfaces with the external solution. I find it revealing that TS15 so far in the article has not at all mentioned such interfaces.
Moreover, in the above sentence TS15 causally states that the anion exclusion process is “observed in diffusion experiments”, without further clarification. Given that the previous section treated diffusion, a reader would expect to have been introduced to the anion exclusion process and how it is observed in diffusion experiments. But this subsection is the first time in TS15 where the term “anion exclusion” is used! In the section on diffusion, “anion accessible porosity” was briefly mentioned, and I suppose a reader is here presumed to connect the dots. But the presence of an exclusion process certainly does not imply an “anion accessible porosity”! Furthermore, anion exclusion is not necessarily observed in diffusion experiments. A more correct statement is that we observe effects of salt exclusion in experiments where a bentonite sample is contacted with an external solution via a semi-permeable component (which typically is a filter that keeps the clay in place). The effect is most conveniently studied in equilibrium rather than diffusion tests, and salt exclusion is not present in e.g. closed-cell diffusion tests. Note that exclusion effects are always related to an external solution.
As the ionic strength increases, the EDL thickness decreases, with
the result that the anion accessible porosity increases as well.
Here it is fully clear that TS15 conflate “anion accessible porosity” and “anion exclusion”. If we consider the “Schofield type” of salt exclusion, it is true that the so-called “exclusion volume” changes with the ionic strength. However, an exclusion volume is not a physical space, but an effective, equivalent quantity. It is derived from the Gouy-Chapman model, which always has anions present everywhere.
Even more importantly, the “Schofield type” of exclusion is not really of interest in dense systems (nor is the Gouy-Chapman model valid in such systems). As discussed above, one must instead consider salt exclusion stemming from charge separation at interfaces with the external solution. For this case it does not even make sense to define an exclusion volume.
I can only interpret this entire paragraph as another fruitless attempt to motivate a multi-porous modeling approach. In this subsection we have so far been told that “two distinct types of porosity” can be defined (they cannot), and we have vaguely been hinted that “bulk or free solution” also is relevant for modelling compacted bentonite. And with the last quoted sentence it is relatively clear that TS15 try to establish that the relative sizes of various “porosities” are controlled by a simple parameter (ionic strength).
The final paragraph of this subsection contain several statements that
makes my jaw drop.
An equivalent anion accessible porosity can be estimated from the
integration of the anion concentration profile (Fig. 6) from the
surface to the bulk water (Sposito 2004)
Here the authors suddenly use the phrase “equivalent”! They are thus obviously aware of that “anion accessible porosity” is a spurious concept?! ?!?! I really don’t know what to say. Their own graph (“Fig. 6”) even show that the Gouy-Chapman model has anions (salt) everywhere! Note that this statement also implies that “the bulk water” is assumed to exist within the clay.
In compacted clay material, the pore sizes may be small as compared
to the EDL size. In that case, it is necessary to take into account
the EDLs overlap between two neighbouring surfaces.
I think this is a very revealing passage. The conditions of compacted bentonite are treated as an exception: pore sizes “may” be smaller than the EDL, and “in that case” it is necessary to account for overlapping diffuse layers. But for compacted bentonite, this is the only relevant situation to consider! Without “overlapping” diffuse layers there is no swelling and no sealing properties. An entire page has been devoted to discussing a model only relevant for suspensions (Gouy-Chapman), while “compacted clay material” here is commented in two sentences…
Clay mineral particles are, however, often segregated into
aggregates delimiting inter-aggregate spaces whose size is usually
larger than inter-particle spaces inside the aggregates.
All of a sudden — in the middle of a paragraph — we are introduced
to a new structural component! “Aggregates” have not been mentioned
earlier in the article and is here introduced without any references.
It is my strong opinion that this way of writing is not appropriate
for a scientific publication, especially not for a review article.
I’m not sure what type of system the authors have in mind here, but
“aggregates” are typically not present in actual water saturated
bentonite. I have commented more on this in
the blog post on stacks.
Conclusion
At the end of the previous section (on diffusion), we were promised that this section should qualitatively link “fundamental properties of the clay minerals” to the diffusional behavior of compacted bentonite. Instead, we are given a fictional description of the structure (conflated with structures of other “clay minerals”), along with a confused explanation of anion exclusion that is irrelevant for such systems. Not a single word is said about the equilibrium that must be considered, namely that at interfaces between bentonite and external solutions. Rather, the idea of “overlapping” diffuse layers — which is the ultimate cause for bentonite swelling — is treated as an exception and only commented on in passing (and nothing is said about how to handle such systems). Although nothing is fully spelled out, I can only interpret this entire part as a (failed) attempt to motivate a multi-porous approach to modeling bentonite. And multi-porosity models cannot be taken seriously.
I admit that scrutinizing studies and pointing out flaws can be fun. However, considering that the descriptions in TS15 are the rule rather than the exception in contemporary bentonite research, I mostly feel weary and resigned. I don’t mean that every clay researcher must agree with me that a homogeneous model is the only reasonable starting point for describing compacted bentonite, and I could only wish that this blog was more influential. But I feel almost dizzy thinking about how this research sector is so hermetically sealed that one can spend entire careers in it without ever having to worry about understanding the nature of swelling and swelling pressure.
Update (250901): Part III of this review is found here.
Footnotes
[1] The Wikipedia article on illite, for example, states that the cation exchange capacity is typically 0.2 — 0.3 eq/kg. Is a significant cation exchange capacity required for classifying something as illite?
[2] E.g. (Poinssot et al., 1999), that TS15 reference as a source on illite, work with sodium exchanged “illite du Puy”, i.e. “Na-illite”.
[3] The material was dispersed by diluting it in alkaline solution and sonicating it. It was thereafter dropped as a suspension on a glass slide and dried.
[4] We may note that the number 5 — 20 TOT-layers in a stack actually showed up when we investigated how this concept is (mis)used in descriptions of bentonite. There it turned out to be a complete misunderstanding of the behavior of suspensions of Ca-montmorillonite.
[5] I am not capable to produce anything reasonable in 3D,
but I think a 2D representation still conveys the message.
[6] Perhaps this criticism can be regarded as nitpicking. I have a nagging feeling, though, that electrostatics is quite poorly understood in certain parts of the bentonite research field. Take the phrase “negative electrostatic potential field”, for example. Although it can be understood at face value (a scalar field with negative values), it also appears to mix together stuff related to charges (“negative”), electric fields (“field”), and potentials. It certainly is important to separate these concepts. There are many examples in the clay literature when this is not done. E.g. Madsen and Müller-Vonmoos (1989) mean that two “potential fields” can repel each other (and also misunderstand swelling)
A high negative potential exists directly at the surface of the
clay layer. […] When two such negative potential fields overlap,
they repel each other, and cause the observed swelling in clay.
[…] the net negative electrical potential
between closely spaced clay particles repel anions attempting to
migrate through the narrow aqueous films of a compact clay […]
In this case, when the clay is compressed […] to the extent that
the electrostatic (diffuse double) layers surrounding the
particles overlap, the overlapping negative potentials repel
invading anions such that the pore becomes excluded to the anion.
[7] An isolated layer of negative charge of course also has an electric field directed towards it, but this is not the relevant system to consider here. (Such a system will actually have an electric field strength that is independent of the distance to the surface, as long as the layer can be regarded as infinitely extended.)
It should go without saying that modelers and model developers must justify every feature, mechanism, or component that they use. Failing to do so strongly increases the risk of being fooled by overparameterization rather than gaining insight. The bentonite scientific literature is nonetheless full of incorrect or unjustified model assumptions, several of which have been discussed previously on the blog. Examples include assuming the presence of bulk water, assuming “stack” structures, and assuming that diffusive fluxes from separate domains are additive. Here we discuss yet another unjustified common model component: Stern layers on montmorillonite basal surfaces.
In the bentonite literature, a Stern layer essentially means a layer of “specifically” sorbed ions on the basal surface, as e.g. illustrated here, in a a figure very similar to what is found in Leroy et al. (2006). Illustrations like this are ubiquitous in the literature.
If montmorillonite basal surfaces function roughly as uniform planes of charge we expect the counter-ions to form a diffuse layer, as e.g. described by the Gouy-Chapman model. By introducing a Stern layer, however, many bentonite researchers mean that exchangeable cations in general also interact with basal surfaces by forming immobile surface complexes. Such interactions necessarily involve mechanisms more “chemical” than the pure electrostatic interaction with uniform planes of charge, and the typical description postulates localized “sorption sites”, as illustrated above.
This blog post treats three different main arguments against Stern layers, presented in different sections
I want to make clear that this criticism concerns one particular type
of surface: the montmorillonite basal surface.
Stern layer models are found in many research fields dealing with
solid interfaces, and although they have been criticized more
generally, here we have no intention of doing so. Likewise, the
process of surface complexation is certainly important generally —
even in bentonite, e.g. on edge surfaces of montmorillonite
particles.1
To better be able to criticize the use of Stern layers on basal surfaces in bentonite modeling, we begin by discussing the origin of Stern’s model.
Origins of the Stern Layer model
The Stern layer concepts were introduced by Otto Stern2 as an extension of the Gouy-Chapman model. Stern’s main concern was metallic electrodes in electrochemical applications. In such systems, the surface (electrode) potential is externally controlled, and can typically be on the order of 1 volt. It is easily seen that the Gouy-Chapman model predicts nonsense for such surface potentials. For e.g. a 1:1 system, the counter-ion concentration at the surface is enhanced by a factor of the order of \(10^{17}\)(!), as seen directly from the Boltzmann distribution \(c^\mathrm{surf} = c^\mathrm{ext}\cdot e^{e\psi^0/kT}\), where \(\psi^0\) is the surface potential, \(e\) is the elementary charge, \(kT\) the thermal energy, and \(c^\mathrm{ext}\) is the concentration far away from the surface. The main problem is that the Gouy-Chapman model does not account for the finite size of ions, and therefore can accumulate an arbitrary amount of charge at the surface. To remedy this flaw, Stern suggested to divide the interface region into a “compact” layer and a “diffuse” part, with the division located an ionic radius from the electrode surface (sometimes referred to as the outer Helmholtz plane).
In the simplest version of Stern’s model the compact layer is free of charges but act as a plate capacitor with a prescribed capacitance (per unit area) \(K_0\). In the original paper Stern shows that, with electrode potential \(\psi^0 = 1\) V and external 1:1 solution concentration \(c^\mathrm{ext} = 1\) M, such a capacitive layer reduces the potential where the diffuse layer begins to \(\psi^1 = 0.08\) V; lowering the external concentration to \(c^\mathrm{ext} = 0.01\) M gives \(\psi^1 = 0.18\) V. For these calculations, Stern uses a value of \(K_0 = 0.29\;\mathrm{F/m^2}\), adopted from measurements on mercury electrodes. This version of the Stern model is essentially a way to take into account that ions cannot get arbitrarily close to the surface.
Stern also presented more elaborate versions of the model that include adsorption in the compact layer (as a Langmuir adsorption model). It is such mechanisms that is universally referred to as a Stern layer in the bentonite scientific literature. Clearly, such versions are substantially more conceptually complex; rather than to just account for a finite ion size at the first molecular layer, we must now also consider additional chemical interactions that typically are different for different types of ions. We also need to have an idea about the adsorption capacity.
Lack of a coherent description of “specific sorption” on
montmorillonite basal surfaces
When using Stern layers for describing montmorillonite basal surfaces, a first thing to note is that the surface potential is not independently controlled for these systems. In contrast to metallic electrodes, montmorillonite is characterized by a fixed surface charge and the problem of accumulating unrealistically large amounts of ions at the interface is significantly mitigated. As pointed by e.g. Norrish and Bolt already in the 1950s, even if we put all counter-ions within the first nanometer adjacent to the surface, the corresponding ion concentration is not larger than approximately 3 M. Here is an illustration of the montmorillonite basal surface on the nm scale, with a representative number of monovalent counter-ions (top layer oxygen atoms are red and the counter-ions blue).3
Clearly, there is room to accommodate all ions without running into the problems that was initially addressed by Stern’s model. Of course, solely accounting for the finite size of the ions — as is done in the simplest version of Stern’s model — is always well justified and will in principle improve the description. In particular, a pure diffuse layer model overestimates the capacitance of the surface. As shown in the table below, the introduction of an empty Stern layer “fixes” this problem.
Here \(c^\mathrm{ext}\) is the concentration of the 1:1 salt far away from the surface, \(\psi^1\) and \(c^1\) denote the electrostatic potential and the counter-ion concentration, respectively, at the point where the diffuse layer begins (i.e. at the interface to the compact layer), and \(\psi^0\) is the electrostatic potential at the surface. \(K_\mathrm{Stern}\) denotes the corresponding capacitance as calculated from the Stern model, with the choice \(K_0 = 0.29 \;\mathrm{F/m^2}\). \(K_\mathrm{DL}\) is instead the capacitance as calculated from a pure diffuse layer model (in which case the surface potential has the value of \(\psi^1\)). In the calculations are assumed a montmorillonite surface charge of 0.111 \(\mathrm{C}/\mathrm{m ^2}\).
But to simply account for the finite size of the ions by means of an empty compact layer is not how the term Stern layer is used in the bentonite scientific literature. As mentioned, most bentonite researchers mean that parts of the rather sparse collection of ions on the surface interacts chemically (“specific sorption”, “chemisorption”). The question of whether Stern layers on montmorillonite basal surfaces are well motivated thus reduces to what arguments there are for more elaborate chemical mechanisms being active on these surfaces. And descriptions of specific sorption on basal surfaces are really all over the place.
Deshpandeand Marshall (1959, 1961) claim that counter-ions are partitioned between (i) chemisorbed ions, which do not contribute to conductivity or activity, (ii) physisorbed ions in a Stern layer, which do not contribute to activity or D.C. conductivity, and (iii) diffuse layer ions, which contribute fully to activity and conductivity. If I interpret their numbers correctly, they state that about 75% — 80% of the counter-ions in pure K-montmorillonite are immobilized. Note that these authors mean that ions in the Stern layer are “physically” adsorbed, while the surface also has “chemically” adsorbed species. Thus, they use the term Stern layer for certain types of physisorption, while stating that ions also bond covalently to the surface.
ShainbergandKemper (1966, 1967), on the other hand, model ions as either mobile in a diffuse layer or immobile in a Stern layer. They argue that covalently bound ions are “extremely unlikely”, and mean that ions in the Stern layer form “ion pairs” with the surface, as suggested earlier by Heald et al. (1964). They use this idea as a starting point for analyzing differences in exchange selectivity for different monovalent cations in montmorillonite. They claim that “about 20 to 50% of sodium are specifically adsorbed”.
Note that Shainberg and Kemper, just like Deshpande and Marshall,
assume Stern layer ions to be “physically” bonded
to the surface (i.e. non-covalently), while having a completely
different opinion on the presence of chemisorbed
ions. Shainberg and Kemper (1966) provide a picture showing the conceptual
difference between an ion in the Stern layer (“unhydrated”) and ions
in the diffuse layer (“hydrated”) that looks very similar to this
In this context it may be worth to also mention the work of
Low
and co-workers. Low
argued consistently that swelling pressure is not primarily related to
the exchangeable ions — something that I strongly disagree with and
that I commented briefly on in a
previous blog post.4 Directly related to this view, these authors
claim that the exchangeable ions for the most part do not dissociate
from the surfaces, and in laterpapers they refer
to such ions as being part of a Stern layer.
To me, all the above descriptions seem like little more than speculation. None of these authors discuss how or why e.g. sodium ions (!) are supposed to from ion pairs with a charge center buried far inside the montmorillonite layer, nor how or why they bond covalently with the basal surface. Nevertheless, both Low as well as Shainberg and Kemper seem to have influenced the writings of Sposito, who, in turn, has had quite a huge impact on contemporary descriptions of bentonite. In e.g. Sposito (1992), which specifically discusses montmorillonite (“smectite”), he writes
Despite the long history of continual investigation of the surface
and colloid chemistry of smectites (van Olphen, 1977; Sposito,
1984), the structure of the electrical double layer at smectite
surfaces and its influence on the rheological properties of smectite
suspensions remain topics of lively controversy. One of the most
contentious issues is the partitioning of adsorbed monovalent
cations among the three possible surface species on the basal planes
of smectite particles, such as montmorillonite (see, e.g., Low,
1981, 1987). […] [A] monovalent cation can be
adsorbed on the basal planes by three different mechanisms:
inner-sphere surface complexes, in which the cation
desolvates and is captured by a ditrigonal cavity;
outer-sphere surface complexes, in which the cation remains
solvated but still is captured by a ditrigonal cavity and
immobilized; and the diffuse-ion swarm, in which the cation
is attracted to the basal plane, but remains fully dissociated from
the smectite surface (Sposito, 1989a, Chap. 7).
The view conveyed here is that exchangeable ions do not interact with montmorillonite basal surfaces as if these, to a first approximation, are planes of uniform charge. Such interaction is only supposed to govern an outer diffuse layer (called a “diffuse-ion swarm” for unclear reasons), and ions are also supposed to interact with the surface by no less than two other “mechanisms”, related to the “ditrigonal cavities”.
Note that while Sposito acknowledges an ongoing “lively controversy” regarding how to describe montmorillonite basal surfaces, he specifies that this debate is limited to how to distribute the exchangeable ions among “three possible surface species.” But, as we will explore below, there is certainly no consensus within colloid chemistry that exchangeable ions are involved in complexation chemistry on the basal surfaces! (I therefore find this way of formulating the “controversy” quite dishonest, to be honest.) For reasons I can’t get my head around, descriptions of “inner-” and “outer-sphere complexes” on montmorillonite basal surfaces are anyway ubiquitous in modern bentonite literature. Let’s therefore take a closer look at how these are introduced.
“Inner-” and “outer-sphere” surface complexes
A description that hardly enlightens me is given in Sposito (1989)5
[The inner- and outer-sphere complexes]
constitute the Stern layer on an adsorbent.
[…]
The diffuse-ion swarm and
the outer-sphere surface complex mechanisms of adsorption involve
almost exclusively electrostatic bonding, whereas inner-sphere
complex mechanisms are likely to involve ionic as well as covalent
bonding. Because covalent bonding depends significantly on the
particular electron configurations of both the surface group and the
complexed ion, it is appropriate to consider inner-sphere surface
complexation as the molecular basis of the term specific
adsorption. Correspondingly, diffuse-ion screening and outer-sphere
surface complexation are the molecular basis for the term
nonspecific adsorption. Nonspecific refers to the weak dependence on
the detailed electron configurations of the surface functional group
and adsorbed ion that is to be expected for the interactions of
solvated species.
Here, Sposito means that exchangeable ions bond covalently with the
montmorillonite basal surface,6 in agreement with Deshpende and Marschall, and in
disagreement with Shainberg and Kemper (we have one “extremely
unlikely” and one “likely” for covalent bonding…). In contrast to
Deshpende and Marschall, however, Sposito means that these
“inner-sphere” complexes are part of the Stern layer. To confuse
matters even more, Shainberg and Kemper assume their “unhydrated”
construct (which corresponds structurally to an “inner-sphere”
complex, see above figure) as being part of the Stern
layer, but not part of any covalent bonding. Moreover,
Shainberg and Kemper assume their “hydrated” construct
(corresponding structurally to an “outer-sphere” complex, see
above figure) to be part of the diffuse layer, while
Sposito wants his “outer-sphere” complexes to be immobile and part
of the Stern layer…
Given the above description (and others) it is hard to understand what
the difference is supposed to be between an “outer-sphere” complex
and an ion in the “diffuse-ion swarm”, other than that the former is
simply claimed to be immobilized; both ions are said to interact with
“exclusively electrostatic bonding”,7 both are classified as “nonspecific
adsorption”, and both are fully hydrated. In my head, this is simply
a recipe for achieving an overparameterized model description.
Sposito’s description also makes implicit statements about the montmorillonite basal surface: it contains “surface functional groups” whose specific electron configuration significantly influence covalent bonding, while being insensitive for the formation of “outer-sphere” complexes and the “diffuse ion-swarm”. In Sposito (1984) he suggests that the “functional groups” are groups of oxygen atoms on the surface of the montmorillonite particle (“ditrigonal cavities”) that qualifies as Lewis bases. The presence of atomic substitutions in the octahedral layer is supposed to enhance this Lewis base character
If isomorphic substitution of \(\mathrm{Al}^{3+}\) by
\(\mathrm{Fe}^{2+}\) or \(\mathrm{Mg}^{2+}\) occurs in the octahedral sheet,
the resulting excess negative charge can distribute itself
principally over the 10 surface oxygen atoms of the four silica
tetrahedra that are associated through their apexes with a single
octahedron in the layer. This distribution of negative charge
enhances the Lewis base character of the ditrigonal cavity and makes
it possible to form complexes with cations as well as with dipolar
molecules.
Note how completely different this whole description is compared to
the original Gouy-Chapman conceptual view. Here is implied that
montmorillonite basal planes8cannot be described as a passive layer of charge,
but that it is a fully reactive system, including covalent
bonding mechanisms.
Frankly, I dismiss the above description of the montmorillonite surface as a Lewis base as pure speculation. I will gladly admit that I am a physicist rather than a chemist, and perhaps I am missing something obvious, but I really don’t see any argumentation behind this description. I am also under the impression that montmorillonite basal planes are relatively chemically stable — that is why they form in the first place, and that is also one reason for why we are interested in using bentonite for e.g. long-term geological waste storage. Furthermore, a meta-argument for dismissing this description is that in later publications we find statements like this, in Sposito (2004):
The \(\mathrm{Na}^+\) that are counterions for the negative structural
charge developed as a result of isomorphic substitutions within the
clay mineral layer tend to adsorb as solvated species on the basal
plane (a plane of hexagonal rings of oxygen ions known as a siloxane
surface) near deficits of negative charge originating in the
octahedral sheet from substitution of a bivalent cation for
\(\mathrm{Al}^{3+}\). This mode of adsorption occurs as a result of
the strong solvating characteristics of Na and the physical
impediment to direct contact between Na and the site of negative
charge posed by the layer structure itself. The way in which this
negative charge is distributed on the siloxane surface is not well
known, but if the charge tends to be delocalized there, that would
also lend itself to outer-sphere surface complexation.
So, 20 years after the surface chemistry of montmorillonite was described as if it was completely understood (Sposito, 1984), the way the negative charge distributes is now described instead as “not well known”… Furthermore, in contrast to earlier statements, the formation of an “outer-sphere complex” is here associated mainly with the hydration properties of sodium. But if the idea of a “surface functional group” is discarded — or at least downplayed — why should a hydrated ion near the surface be described as a surface complex at all?
We note that Sposito (2004) still seem to imply that the “outer-sphere surface complex” is localized and immobile (“adsorbed near deficits of negative charge”) But the evidence is vast that sodium, and several other ions, are quite mobile even in monohydrates (see below).
Deviations from the Gouy-Chapman model do not imply surface complexation
Authors that promote Stern layers on montmorillonite basal surfaces
usually rely on the Gouy-Chapman model for describing the diffuse
layer part. Lyklema,
writing generally on colloid science, explicitly “defends” such an
approach
In the following our discussion will be based on the rather
pragmatic, though somewhat artificial, subdivision of the solution
side of the double layer into two parts: an inner part, or
Stern layer where all complications regarding finite ion
size, specific adsorption, discrete charges, surface heterogeneity,
etc., reside and an outer, Gouy or diffuse
layer, that is by definition ideal, i.e. it obeys
Poisson-Boltzmann statistics. This model is due to Stern following
older ideas of Helmholtz and has over the decades since its
inception rendered excellent services, especially in dealing with
experimental systems.
Bolt and co-workers […] investigated in detail the application of
the Gouy–Chapman diffuse-layer theory to ion-exchange
processes. Their work demonstrated that consideration of
electrostatic sorption alone is not sufficient to explain
ion-exchange data and that chemisorption (or “specific” sorption)
needs to be included in ion-exchange models.
It is not logically consistent to conclude that deviations from the
Gouy-Chapman model implies that specific sorption “needs to be
included”.9 On the contrary, introducing
specific sorption to compensate for a certain model rather than for
surface chemical reasons may, in my mind, be a recipe for an
overparameterized disaster. I don’t get reassured by statements like
this, also from Dzombak and Hudson (1995)
Surface complexation models can be extended to include diffuse-layer
sorption. This approach permits their application in modeling the
sorption of ions (such as monovalent electrolyte ions) that exhibit
weak specific sorption. The generality of such an extended surface
complexation approach together with the mathematical power of modern
chemical speciation models offers the potential for accurate
physicochemical modeling of ion exchange
Reasonably, a complex system may require complex models, but it is
certainly dangerous in a modeling context to rely too heavily on
“mathematical power” (I guess “numerical power” is the preferred
phrase).10
Note that very different attitudes towards the Stern layer concept is found in the colloid science literature, where e.g. Evans and Wennerström (1999) describe it as an “intellectual cul de sac”.11
One way of dealing with these difficulties is to say that the
solution layer closest to a charged surface has properties so
different from the bulk that it should be treated as a separate
entity. This device was introduced in the 1930s by the German
electrochemist Stern and the surface layer is commonly referred to
as the Stern layer, whose properties are specified by a number of
empirical parameters. It is the opinion of the authors of this book
that the Stern layer concept is an intellectual cul de sac for the
description of electrostatics in colloidal systems. One reason for
this point of view is that from modern spectroscopic measurements we
know molecular properties are not dramatically changed for a liquid
close to a charged surface.
I find it quite perplexing that so many authors in the bentonite scientific community attribute any deviation from the Gouy-Chapman model solely to surface-related mechanisms. The Gouy-Chapman model treats both ions (point particles) and water (a dielectric continuum) in a very simplified manner, and it is clear that “specific ion” effects are ubiquitous, also in systems that lack surfaces. Addressing differences in e.g. selectivity coefficients without considering ion polarizability and hydration, while postulating the existence of localized sorption “sites”, can, to my mind, only lead to incorrect descriptions.
The Poisson-Boltzmann equation is a mean field
approximation
Note also that the Poisson-Boltzmann equation — which underlies the Gouy-Chapman model — is only approximate. It is derived by assuming that the electrostatic potential experienced by any ion is the average potential from all other ions (and surfaces). More accurately, the ion distribution around a given ion deviates from the average, as a direct consequence of the presence of the central ion.
Including these ion-ion correlation effects makes the mathematical description considerably more complex. But with the advent of sufficiently powerful computers and algorithms, the electric double layer has been solved basically “exactly”. The “exact” solution may differ strongly from the Poisson-Boltzmann solution, with increasing concentrations towards the surfaces (and consequently a lowering of interlayer midpoint concentrations), and an explicit attractive electrostatic force between the two halves of an interlayer. Using Monte Carlo simulations, Guldbrand et al. (1984) demonstrated that with divalent counter-ions these effects are so large that the system becomes net attractive at a certain interlayer distance, in qualitative disagreement with the Poisson-Boltzmann solution. This effect, which has been thoroughlystudiedsincethe 80s, and which we have discussed in severalposts on this blog, is the prevailing explanation e.g. for the limited swelling of Ca-montmorillonite.
The lesson here is that observed deviations from predictions of the Poisson-Boltzmann equation not automatically can be taken as evidence for additional active system components, and certainly not as evidence for specific sorption. Note that limited swelling in divalent montmorillonite is explained by the ions being diffusive, not that they are sorbed and immobilized. I cannot overstate the importance of this insight.
It boggles my mind that the entire research area on ion-ion correlations in colloidal systems seems to have made no significant impact on parts of the bentonite scientific community; I seldom find references to works on ion-ion correlation, and when I do it’s quite confused. E.g. Sposito (1992) means that the formation of “quasicrystals”12 is due to “outer-sphere” complexes
The best known example of a montmorillonite quasicrystal is that
comprising stacks of four to seven layers. \(\mathrm{Ca}^{2+}\) ions,
solvated by six water molecules (outer-sphere surface complex),
serve as molecular “cross-links” to help bind the clay layers
together through electrostatic forces.
Sure, the ion-ion correlation effect that prevents Ca-montmorillonite from exfoliating is of electrostatic origin, but it is not related to “cross-links” or surface complexes. Sposito furthermore continues by claiming that “even […] Na-montmorillonite” forms “quasicrystals”. Such claim cannot be supported by ion-ion correlation — on the contrary, ion-ion correlation explains why Ca-montmorillonite forms “quasicrystals”, while Na-montmorillonite does not. It is thus relatively clear that Sposito do not refer to ion-ion correlation in the above statement. At the same time, later in the same publication he cites Kjellander et al. (1988) on going beyond the mean-field treatment of the Poisson-Boltzmann equation, and even claims that the Gouy-Chapman model is “completely inaccurate” for systems containing divalent ions. I can only conclude that this is a quite confused description.
Finite-size effects of water molecules
With focus on the first molecular layers at a solid interface, it is
clear that finite-size effects of water molecules — which are
not treated in the Gouy-Chapman model — reasonably influences
the resulting ion distribution. This influence is manifested both as a
steric effect — there can only be a discrete number of water
molecules between the ion and the surface — and as an effect of how
strongly a certain ion is hydrated.
Water molecules are treated explicitly e.g. in molecular dynamics (MD) simulations of montmorillonite/water interfaces, and here are results from simulating a three water-layer interlayer of Na-montmorillonite, from Hedström and Karnland (2012)13
Sodium is seen to accumulate “between” the water layers; in the above illustration we have also included schematic illustrations of the molecular configurations, as conceived by Shainberg and Kemper (1966) (shown earlier). As stated earlier, Shainberg and Kemper (1966) refer to these as “hydrated” and “unhydrated”, but they are clearly the same type of configurations that e.g. Sposito (1992) and Dzombak and Hudson (1995) call “outer-” and “inner-sphere” complexes.
While the above mentioned authors mean that these “complexes” involve specific interactions between ions and surface, the MD simulation suggests that such structures are mainly a consequence of the finite-size of the molecules and ions. In particular, the MD results do not support the idea that these structures depend critically on a specific, non-electrostatic, ion–surface interaction. Indeed, the simulations explicitly treat also the atoms of the montmorillonite layer, which could make it difficult to judge whether the appearance of “complexes” mainly is related to water–ion or ion–surface interactions. But note that Hedström and Karnland (2012) simulate two different systems: one where the montmorillonite charge is put on specific atoms in the octahedral sheet (Mg for Al substitutions), and one where it is distributed on all Al atoms (as a fraction of the elementary charge). Both systems have essentially an identical atomic configuration in the interlayer, which strongly suggest that no critical ion–surface interaction is involved in forming “outer-” and “inner-sphere complexes” (i.e. they really are not surface complexes). I am not aware of any published simulation where the basal surface is represented as a uniform sheet of charge while water molecules are treated explicitly, but I am convinced that “outer-” and “inner-sphere complexes” would appear also in such a simulation.
Regarding MD simulations of montmorillonite interlayers, you can also
simply observe them to convince yourself that the counter-ions
are not in any reasonable sense immobilized. These types of
simulations
are routinely used to calculate (quite significant) interlayer diffusion
coefficients, for crying out loud!
Experimental evidence of counter-ion mobility
A final argument for why Stern layers on montmorillonite basal
surfaces are unjustified is the vast amount of empirical evidence of
counter-ion mobility. We have discussed
severaldiffusion studies in
earlierblogposts that show that many ions (Na, Cl, K, Sr, I, Cs, Ca,…) have
a significant mobility even in very dense systems, dominated by bi- or
monohydrated interlayers. In
the previous post, we brought up the following result
This figure shows the resulting concentration profiles in two
diffusion experiments where sodium and chloride tracers, respectively, have diffused from an initial planar
source for the same amount of time (23.7 h), in samples of pure
Na-montmorillonite of dry density 1.8 \(\mathrm{g/cm^3}\), equilibrated
with deionized water. This result was used previously to dismiss the
ludicrous idea that these two ions are supposed to migrate in separate
parts of the pore volume, exposed to completely different
mechanisms. In the same vein, this result can be used to dismiss the
idea of a Stern layer on basal surfaces.
Sodium, which is universally acknowledged to reside in the interlayers,
is here demonstrated to diffuse just fine in bi- and monohydrated
interlayers. As chloride, which also resides in the interlayers
(despite all talk of
“anion-accessible porosity”), behave essentially identical, it is
quite far-fetched to assume any significant surface complexation
mechanism. And anyone who argues for that these tracers actually do
not diffuse in the interlayers should be reminded of the
seeming “uphill”diffusion experiment,14 which is performed at even higher density, and where
the “uphill” diffusion direction once and for all proves that the
transport occurs in interlayers.
Strangely, manyauthorsnowadays seem to promote both Stern layers and interlayer mobility in bentonite. Various simulation codes has been modified for this possibility, and there are several examples of researchers pointing out a possibility of “Stern layerdiffusion”. I think these authors should carefully examine their chain of assumptions: Surface complexation in a Stern layer (i.e. sorption) is initially suggested to explain e.g. why breakthrough times in cation through-diffusion tests are relatively long as compared with the steady-state flux (i.e. why “\(D_e\)” can be considerably larger than “\(D_a\)”). With evidence for that the “sorbed” ions actually dominate the mass transfer, the sorption mechanism is not reconsidered, but yet another mechanism is suggested: Stern layer mobility… Reasonably, such an approach is not adequate for developing models; researchers employing it should critically consider the intended purpose of a Stern layer component.
Counter-ion mobility is also related to swelling pressure. Bentonite swelling pressure is difficult to describe generally, and I have written a whole series of blog posts on the subject, but it is clear that measured swelling pressures in e.g. moderately dense Na- and Li-montmorillonites is quite well described by the Poisson-Boltzmann equation. As this set of conditions (not too dense clay, simple monovalent ions) are exactly those for which we expect the Poisson-Boltzmann equation to be adequate, this is a strong indication that all counter-ions contribute to the pressure.15 Also, the limited swelling in e.g. Ca-montmorillonite, as previously discussed, is explained by ion-ion correlation effects where all ions are included in the diffuse layer.
Finally, we can take a look at salt exclusion from compacted
bentonite. The magnitude of salt exclusion is
directly related to the amount of mobile counter-ions. Thus, if most
of the counter-ions were immobilized in a Stern layer, bentonite
should show small exclusion effects. In contrast, the empirical
results for e.g. chloride exclusion in sodium dominated bentonite
indicate, again, that all counter-ions are part of a diffuse
layer.
This diagram shows the relative amount of chloride in the bentonite as a function of \(c^\mathrm{ext}/c_\mathrm{IL}\), where \(c^\mathrm{ext}\) is the external salt concentration and \(c_\mathrm{IL}\) is the amount exchangeable cations, expressed as a monovalent interlayer concentration. The experimental data is from Van Loon et al. (2007), which we reevaluated and examined in detail in a previous blog post. The lines are the result from applying the “ideal” Donnan formula with various amounts of the counter-ions assumed diffusive. For details on Donnan theory, see this blog post.
Although the experimental data show considerable scatter, there is nothing in this plot that suggests that a fraction of the counter-ions are immobilized. And the quality of this data is certainly good enough to directly dismiss models that assume that the major part of ions are immobilized in a Stern layer.
[1] I find it quite frustrating that many descriptions
in the literature only refer abstractly to “mineral surfaces”
rather than specifically addressing montmorillonite. At the same
time it is often clear from the context that statements regarding
“mineral surfaces” should be understood as applicable to
montmorillonite basal surfaces. I would much appreciate if
researchers promoting Stern layers on basal surfaces would provide
descriptions for specific systems, e.g. pure Na-Ca-montmorillonite.
[2] Otto Stern is a fascinating character in the history of science, most famous for the Stern-Gerlasch experiment, that helped pave the way for quantum mechanics. I highly recommend this lecture by the late Sandip Pakvasa. An example of its contents:
A note on Stern’s style of working: He always had a cigar in one hand, and he left actual work with hands to others, as he did not trust his own manual dexterity! […] He described the beneficial effects of a large wooden hammer that he kept in his lab and used it to threaten the apparatus if it did not behave! (apparently it worked!)
[3] Note that di-valent counter-ions
would be even more sparsely distributed than this.
[4] It may be worth discussing my
objections against the work of Low and co-workers in more detail in
a future blog post.
[5] This is a general discussion on sorption on mineral surfaces, and is cited from the second edition of the book (2008). There is also a “Thired” edition.
[6] This particular description is
general (for “adsorbents”), but since Sposito, as well as a large
part of the contemporary bentonite scientific community, claim that
“inner-sphere” complexes are present on montmorillonite basal
surfaces, we can conclude that they mean that covalent bonding occur
on such surfaces.
[7] Sure, the full
quotation is “almost exclusively electrostatic bonding”, but what
is a reader supposed to do with that? Such vague and sloppy
scientific writing annoys me.
[8] Again, the discussion is on
general “mineral surfaces”, but from other writings it is clear
that this is supposed to apply to the montmorillonite basal
surface.
[9] I furthermore don’t believe that “Bolt and co-workers” concluded that specific sorption “needs” to be included, but that this rather is an interpretation made by Dzombak and Hudson themselves. Bolt considered and downplayed the Stern Layer already in the mid 50s, and although he indeed has expressed positive attitudes (seriously, these guys just write too much!), he continued to downplay its significance in e.g. Bolt (1979), writing “In conclusion it appears justified to assume that for homoionic clays saturated with common ions, if hydrated, the Stern layer will be an “empty” Stern layer according to the terminology of Grahame (1947).”
[10] Note also that the perspective in this quotation is that specific sorption models can be complemented with diffuse layer features — i.e. the existence of sorption “sites” is assumed a priori. But Dzombak and Hudson (1995) never really discuss the nature of such “sites” on montmorillonite basal surfaces, but rely on Sposito’s speculations about “inner-” and “outer-surface” complexes.
[11] Note that Stern’s original paper actually is from 1924. I also suspect that Stern would object to being labeled an “electrochemist”.
[12] The
terminology here is quite messy, and other authors may use other
terms such as “tactoids”, see
this post for a further discussion.
[14] Anyone making this argument should
also provide a plausible suggestion for where a significant
non-interlayer pore structure is located at these extreme
densities.
[15] The pressure in these types of calculations can be related to the interlayer midpoint concentration. But this does not mean that not all counter-ions are involved in the process.
“Multi-porosity” models1 — i.e
models that account for both a bulk water phase and one, or several,
other domains within the clay — have become increasingly
popular in bentonite research during the last couple of decades. These
are obviously macroscopic, as is clear e.g. from the benchmark
simulations described in
Alt-Epping et
al. (2015), which are specified to be discretized into 2 mm thick
cells; each cell is consequently assumed to contain billions and
billions individual montmorillonite particles. The macroscopic
character is also relatively clear in their description of two
numerical tools that have implemented multi-porosity
PHREEQC and CrunchFlowMC have implemented a Donnan approach to describe the electrical potential and species distribution in the EDL. This approach implies a uniform electrical potential \(\varphi^\mathrm{EDL}\) in the EDL and an instantaneous equilibrium distribution of species between the EDL and the free water (i.e., between the micro- and macroporosity, respectively). The assumption of instantaneous equilibrium implies that diffusion between micro- and macroporosity is not considered explicitly and that at all times the chemical potentials, \(\mu_i\), of the species are the same in the two porosities
On an abstract level, we may thus illustrate a multi-porosity approach
something like this (here involving two domains)
The model is represented by one
continuum for the “free water”/”macroporosity” and one for the
“diffuse layer”/”microporosity”,2 which are
postulated to be in equilibrium within each macroscopic cell.
But such an equilibrium (Donnan equilibrium)
requires a
semi-permeable component. I am not aware of any suggestion for such
a component in any publication on multi-porosity
models. Likewise, the co-existence of diffuse layer and free water
domains requires
a mechanism that prevents swelling and maintains the pressure
difference — also the water chemical potential should of course be
the equal in the two “porosities”.3
Note that the questions of what constitutes the semi-permeable
component and what prevents swelling have a clear answer in
the homogeneous mixture model. This answer also corresponds to an
easily identified real-world object: the metal filter (or similar
component) separating the sample from the external solution.
Multi-porosity models, on the other hand, attribute no particular
significance to interfaces between sample and external
solutions. Therefore, a candidate for the semi-permeable component has
to be — but isn’t — sought elsewhere. Donnan equilibrium
calculations are virtually meaningless without identifying this
component.
The partitioning between diffuse layer and free water in
multi-porosity models is, moreover, assumed to be controlled by water
chemistry, usually by means of the
Debye length. E.g. Alt-Epping et al. (2015) write
To determine the volume of the microporosity, the surface area of montmorillonite, and the Debye length, \(D_L\), which is the distance from the charged mineral surface to the point where electrical potential decays by a factor of e, needs to be known. The volume of the microporosity can then be calculated as \begin{equation*} \phi^\mathrm{EDL} = A_\mathrm{clay} D_L, \end{equation*} where \(A_\mathrm{clay}\) is the charged surface area of the clay mineral.
I cannot overstate how strange the multi-porosity description
is. Leaving the abstract representation, here is an attempt to
illustrate the implied clay structure, at the “macropore” scale
The view emerging from the above description is actually even more
peculiar, as the “micro” and “macro” volume fractions are supposed
to vary with the Debye length. A more general illustration of how the
pore structure is supposed to function is shown in this animation
(“I” denotes ionic strength)
What on earth could constitute such magic semi-permeable membranes?!
(Note that they are also supposed to withstand the inevitable pressure
difference.)
Here, the informed reader may object and point out that no researcher
promoting multi-porosity has this magic pore structure in
mind. Indeed, basically all multi-porosity publications instead
vaguely claim that the domain separation occurs on the nanometer scale
and present microscopic illustrations, like this (this is a
simplified version of what is found in
Alt-Epping et
al. (2015))
In the remainder of this post I will discuss how the idea of a domain separation on the microscopic scale is even more preposterous than the magic membranes suggested above. We focus on three aspects:
The implied structure of the free water domain
The arbitrary domain division
Donnan equilibrium on the microscopic scale is not really a valid concept
Implied structure of the free water domain
I’m astonished by how little figures of the microscopic scale are
explained in many publications. For instance, the illustration above
clearly suggests that “free water” is an interface region with
exactly the same surface area as the “double layer”. How can that
make sense? Also, if the above structure is to be taken seriously it
is crucial to specify the extensions of the various water layers. It
is clear that the figure shows a microscopic view, as it depicts an
actual diffuse layer.4 A diffuse layer width varies, say, in the
range 1 – 100 nm,5 but authors seldom reveal if we are
looking at a pore 1 nm wide or several hundred nm wide. Often we are
not even shown a pore — the water film just ends in a void, as in the
above figure.6
The vague nature of these descriptions indicates that they are merely “decorations”, providing a microscopic flavor to what in effect still is a macroscopic model formulation. In practice, most multi-porosity formulations provide some ad hoc mean to calculate the volume of the diffuse layer domain, while the free water porosity is either obtained by subtracting the diffuse layer porosity from total porosity, or by just specifying it. Alt-Epping et al. (2015), for example, simply specifies the “macroporosity”
The total porosity amounts to 47.6 % which is divided into 40.5 % microporosity (EDL) and 7.1 % macroporosity (free water). From the microporosity and the surface area of montmorillonite (Table 7), the Debye length of the EDL calculated from Eq. 11 is 4.97e-10 m.
Clearly, nothing in this description requires or suggests that the
“micro” and “macroporosities” are adjacent waterfilms on the
nm-scale. On the contrary, such an interpretation becomes quite
grotesque, with the “macroporosity” corresponding to half a
monolayer of water molecules! An illustration of an actual pore of
this kind would look something like this
This interpretation becomes even more bizarre, considering that
Alt-Epping et
al. (2015) assume advection to occur only in this half-a-monolayer
of water, and that the diffusivity is here a factor 1000 larger than
in the “microporosity”.
As another example, Appelo
and Wersin (2007) model a cylindrical sample of “Opalinus clay”
of height 0.5 m and radius 0.1 m, with porosity 0.16, by discretizing
the sample volume in 20 sections of width 0.025 m. The void volume of
each section is consequently
\(V_\mathrm{void} = 0.16\cdot\pi\cdot 0.1^2\cdot 0.025\;\mathrm{m^3} =
1.257\cdot10^{-4}\;\mathrm{m^3}\). Half of this volume (“0.062831853”
liter) is specified directly in the input file as the volume of the
free water;7 again, nothing suggests that this water
should be distributed in thin films on the nm-scale. Yet,
Appelo and Wersin (2007)
provide a figure, with no length scale, similar in spirit to that
above, that look very similar to this
They furthermore write about this figure (“Figure 2”)
It should be noted that the model can zoom in on the nm-scale suggested by Figure 2, but also uses it as the representative form for the cm-scale or larger.
I’m not sure I can make sense of this statement, but it seems that they imply that the illustration can serve both as an actual microscopic representation of two spatially separated domains and as a representation of two abstract continua on the macroscopic scale. But this is not true!
Interpreted macroscopically, the vertical dimension is fictitious, and
the two continua are in equilibrium in each paired cell. On a
microscopic scale, on the other hand, equilibrium between paired cells
cannot be assumed a priori, and it becomes crucial to specify
both the vertical and horizontal length scales. As
Appelo and Wersin (2007)
formulate their model assuming equilibrium between paired cells, it is
clear that the above figure must be interpreted macroscopically (the
only reference to a vertical length scale is that the “free
solution” is located “at infinite distance” from the surface).
We can again work out the implications of anyway interpreting the model microscopically. Each clay cell is specified to contain a surface area of \(A_\mathrm{surf}=10^5\;\mathrm{m^2}\).8 Assuming a planar geometry, the average pore width is given by (\(\phi\) denotes porosity and \(V_\mathrm{cell}\) total cell volume)
The double layer thickness is furthermore specified to be 0.628 nm.9 A microscopic interpretation of this particular model thus implies that the sample contains a single type of pore (2.51 nm wide) in which the free water is distributed in a thin film of width 1.25 nm — i.e. approximately four molecular layers of water!
Rather than affirming that multi-porosity model formulations are macroscopic at heart, parts of the bentonite research community have instead doubled down on the confusing idea of having free water distributed on the nm-scale. Tournassat and Steefel (2019) suggest dealing with the case of two parallel charged surfaces in terms of a “Dual Continuum” approach, providing a figure similar to this (surface charge is -0.11 C/m2 and external solution is 0.1 M of a 1:1 electrolyte)
Note that here the perpendicular length scale is specified,
and that it is clear from the start that the electrostatic potential
is non-zero everywhere. Yet,
Tournassat and Steefel
(2019) mean that it is a good idea to treat this system as if it
contained a 0.7 nm wide bulk water slice at the center of the
pore. They furthermore express an almost “postmodern” attitude
towards modeling, writing
It should be also noted here that this model refinement does not imply necessarily that an electroneutral bulk water is present at the center of the pore in reality. This can be appreciated in Figure 6, which shows that the Poisson–Boltzmann predicts an overlap of the diffuse layers bordering the two neighboring surfaces, while the dual continuum model divides the same system into a bulk and a diffuse layer water volume in order to obtain an average concentration in the pore that is consistent with the Poisson–Boltzmann model prediction. Consequently, the pore space subdivision into free and DL water must be seen as a convenient representation that makes it possible to calculate accurately the average concentrations of ions, but it must not be taken as evidence of the effective presence of bulk water in a nanoporous medium.
I can only interpret this way of writing (“…does not imply
necessarily that…”, “…must not be taken as evidence of…”)
that they mean that in some cases the bulk phase should be
interpreted literally, while in other cases the bulk phase
should be interpreted just as some auxiliary component. It is my
strong opinion that such an attitude towards modeling only contributes
negatively to process understanding (we may e.g. note that later in
the article, Tournassat
and Steefel (2019) assume this perhaps non-existent bulk water to
be solely responsible for advective flow…).
I say it again: no matter how much researchers discuss them in microscopic terms, these models are just macroscopic formulations. Using the terminology of Tournassat and Steefel (2019), they are, at the end of the day, represented as dual continua assumed to be in local equilibrium (in accordance with the first figure of this post). And while researchers put much effort in trying to give these models a microscopic appearance, I am not aware of anyone suggesting a reasonable candidate for what actually could constitute the semi-permeable component necessary for maintaining such an equilibrium.
Arbitrary division between diffuse layer and free water
Another peculiarity in the multi-porosity descriptions showing that they cannot be interpreted microscopically is the arbitrary positioning of the separation between diffuse layer and free water. We saw earlier that Alt-Epping et al. (2015) set this separation at one Debye length from the surface, where the electrostatic potential is claimed to have decayed by a factor of e. What motivates this choice?
Most publications on multi-porosity models define free water as a region where the solution is charge neutral, i.e. where the electrostatic potential is vanishingly small.10 At the point chosen by Alt-Epping et al. (2015), the potential is about 37% of its value at the surface. This cannot be considered vanishingly small under any circumstance, and the region considered as free water is consequently not charge neutral.
The diffuse layer thickness chosen by Appelo and Wersin (2007) instead corresponds to 1.27 Debye lengths. At this position the potential is about 28% of its value at the surface, which neither can be considered vanishingly small. At the mid point of the pore (1.25 nm), the potential is about 8%11 of the value at the surface (corresponding to about 2.5 Debye lengths). I find it hard to accept even this value as vanishingly small.
Note that if the boundary distance used by Appelo and Wersin (2007) (1.27 Debye lengths) was used in the benchmark of Alt-Epping et al. (2015), the diffuse layer volume becomes larger than the total pore volume! In fact, this occurs in all models of this kind for low enough ionic strength, as the Debye length diverges in this limit. Therefore, many multi-porosity model formulations include clunky “if-then-else” clauses,12 where the system is treated conceptually different depending on whether or not the (arbitrarily chosen) diffuse layer domain fills the entire pore volume.13
In the example from Tournassat and Steefel (2019) the extension of the diffuse layer is
1.6 nm, corresponding to about 1.69 Debye lengths. The potential is
here about 19% of the surface value (the value in the midpoint is
12% of the surface
value). Tournassat
and Appelo (2011) uses yet another separation distance — two Debye
lengths — based on
misusing the concept of exclusion volume in the Gouy-Chapman model.
With these examples, I am not trying to say that a better criterion is needed for the partitioning between diffuse layer and bulk. Rather, these examples show that such a partitioning is quite arbitrary on a microscopic scale. Of course, choosing points where the electrostatic potential is significant makes no sense, but even for points that could be considered having zero potential, what would be the criterion? Is two Debye lengths enough? Or perhaps four? Why?
These examples also demonstrate that researchers ultimately do not
have a microscopic view in mind. Rather, the “microscopic”
specifications are subject to the macroscopic constraints.
Alt-Epping et
al. (2015), for example, specifies a priori that the system
contains about 15% free water, from which it follows that the diffuse
layer thickness must be set to about one Debye length (given the
adopted surface area). Likewise,
Appelo and Wersin (2007)
assume from the start that Opalinus clay contains 50% free water, and
set up their model accordingly.14Tournassat and Steefel
(2019) acknowledge their approach to only be a “convenient
representation”, and don’t even relate the diffuse layer
extension to a specific value of the electrostatic
potential.15 Why
the free water domain anyway is considered to be positioned in the
center of the nanopore is a mystery to me (well, I guess because
sometimes this interpretation is supposed to be taken literally…).
Note that none of the free water domains in the considered models are actually charged, even though the electrostatic potential in the microscopic interpretations is implied to be non-zero. This just confirms that such interpretations are not valid, and that the actual model handling is the equilibration of two (or more) macroscopic, abstract, continua. The diffuse layer domain is defined by following some arbitrary procedure that involves microscopic concepts. But just because the diffuse layer domain is quantified by multiplying a surface area by some multiple of the Debye length does not make it a microscopic entity.4
Donnan effect on the microscopic scale?!
Although we have already seen that we cannot interpret multi-porosity models microscopically, we have not yet considered the weirdest description adopted by basically all proponents of these models: they claim to perform Donnan equilibrium calculations between diffuse layer and free water regions on the microscopic scale!
The underlying mechanism for a Donnan effect is the establishment of charge separation, which obviously occur on the scale of the ions, i.e. on the microscopic scale. Indeed, a diffuse layer is the manifestation of this charge separation. Donnan equilibrium can consequently not be established within a diffuse layer region, and discontinuous electrostatic potentials only have meaning in a macroscopic context.
Consider e.g. the interface between bentonite and an external solution
in
the
homogeneous mixture model. Although this model ignores the
microscopic scale, it implies charge separation and a continuously
varying potential on this scale, as illustrated here
The regions where the potential varies are exactly what we categorize
as diffuse layers (exemplified in two ideal microscopic geometries).
The discontinuous potentials encountered in multi-porosity model descriptions (see e.g. the above “Dual Continuum” potential that varies discontinuously on the angstrom scale) can be drawn on paper, but don’t convey any physical meaning.
Here I am not saying that Donnan equilibrium calculations cannot be performed in multi-porosity models. Rather, this is yet another aspect showing that such models only have meaning macroscopically, even though they are persistently presented as if they somehow consider the microscopic scale.
An example of this confusion of scales is found in
Alt-Epping et
al. (2018), who revisit the benchmark problem of
Alt-Epping et
al. (2015) using an alternative approach to Donnan equilibrium:
rather than directly calculating the equilibrium, they model the clay
charge as immobile mono-valent anions, and utilize the
Nernst-Planck
equations. They present “the conceptual model” in a figure very
similar to this one
This illustration simultaneously conveys both a micro- and macroscopic view. For example, a mineral surface is indicated at the bottom, suggesting that we supposedly are looking at an actual interface region, in similarity with the figures we have looked at earlier. Moreover, the figure contains entities that must be interpreted as individual ions, including the immobile “clay-anions”. As in several of the previous examples, no length scale is provided (neither perpendicular to, nor along the “surface”).
On the other hand, the region is divided into cells, similar to the
illustration in Appelo and Wersin (2007). These can hardly have any other meaning
than to indicate the macroscopic discretization in the adopted
transport code (FLOTRAN). Also, as the “Donnan porosity” region
contains the “clay-anions” it can certainly not represent a diffuse
layer extending from a clay surface; the only way to make sense of
such an “immobile-anion” solution is that it represents a
macroscopic homogenized clay domain (a homogeneous mixture!).
Furthermore, if the figure is supposed to show the microscopic scale
there is no Donnan effect, because there is no charge separation!
Taking the depiction of individual ions seriously, the interface
region should rather look something like this in equilibrium
This illustrates the fundamental problem with a Donnan effect between microscopic compartments: the effect requires a charge separation, whose extension is the same as the size of the compartments assumed to be in equilibrium.16
Despite the confusion of the illustration in Alt-Epping et al. (2018), it is clear that a macroscopic model is adopted, as in our previous examples. In this case, the model is explicitly 2-dimensional, and the authors utilize the “trick” to make diffusion much faster in the perpendicular direction compared to the direction along the “surface”. This is achieved either by making the perpendicular diffusivity very high, or by making the perpendicular extension small. In any case, a perpendicular length scale must have been specified in the model, even if it is nowhere stated in the article. The same “trick” for emulating Donnan equilibrium is also used by Jenni et al. (2017), who write
In the present model set-up, this approach was implemented as two connected domains in the z dimension: one containing all minerals plus the free porosity (z=1) and the other containing the Donnan porosity, including the immobile anions (CEC, z=2, Fig. 2). Reproducing instantaneous equilibrium between Donnan and free porosities requires a much faster diffusion between the porosity domains than along the porosity domains.
Note that although the perpendicular dimension (\(z\)) here is referred
to without unit(!), this representation only makes sense in a
macroscopic context.
Jenni et al. (2017) also provide a statement that I think fairly well sums up the multi-porosity modeling endeavor:17
In a Donnan porosity concept, cation exchange can be seen as resulting from Donnan equilibrium between the Donnan porosity and the free porosity, possibly moderated by additional specific sorption. In CrunchflowMC or PhreeqC (Appelo and Wersin, 2007; Steefel, 2009; Tournassat and Appelo, 2011; Alt-Epping et al., 2014; Tournassat and Steefel, 2015), this is implemented by an explicit partitioning function that distributes aqueous species between the two pore compartments. Alternatively, this ion partitioning can be modelled implicitly by diffusion and electrochemical migration (Fick’s first law and Nernst-Planck equations) between the free porosity and the Donnan porosity, the latter containing immobile anions representing the CEC. The resulting ion compositions of the two equilibrated porosities agree with the concentrations predicted by the Donnan equilibrium, which can be shown in case studies (unpublished results, Gimmi and Alt-Epping).
Ultimately, these are models that, using one approach or the other,
simply calculates Donnan equilibrium between two abstract,
macroscopically defined domains (“porosities”,
“continua”). Microscopic interpretations of these models lead — as
we have demonstrated — to multiple absurdities and errors. I am not
aware of any multi-porosity approach that has provided any kind of
suggestion for what constitutes the semi-permeable component required
for maintaining the equilibrium they are supposed to describe.
Alternatively expressed: what, in the previous figure,
prevents the “immobile anions” from occupying the entire clay
volume?
The most favorable interpretation I can make of multi-porosity approaches to bentonite modeling is a dynamically varying “macroporosity”, involving magical membranes (shown above). This, in itself, answers why I cannot take multi-porosity models seriously. And then we haven’t yet mentioned the flawed treatment of diffusive flux.
[1] This category has many other names,
e.g. “dual
porosity” and “dual continuum”, models. Here, I mostly use the term
“multi-porosity” to refer to any model of this kind.
[3] This lack of a full
description is very much related to the incomplete description of
so-called
“stacks” — I am not aware of any reasonable suggestion of a
mechanism for keeping stacks together.
[4] Note the difference between a diffuse layer and a diffuse layer domain. The former is a structure on the nm-scale; the latter is a macroscopic, abstract model component (a continuum).
[5] The scale of an electric double layer is
set by the Debye length, \(\kappa^{-1}\). From the formula for a 1:1
electrolyte, \(\kappa^{-1} = 0.3 \;\mathrm{nm}/\sqrt{I}\), the Debye
length is seen to vary between 0.3 nm and 30 nm when ionic strength
is varied between 1.0 M to 0.0001 M (\(I\) is the numerical value of
the ionic strength expressed in molar units). Independent of the
value of the factor used to multiply \(\kappa^{-1}\) in order to
estimate the double layer extension, I’d say that the estimation 1 –
100 nm is quite reasonable.
[6] Here, the informed reader may perhaps point out that authors don’t really mean that the free water film has exactly the same geometry as the diffuse layer, and that figures like the one above are more abstract representations of a more complex structure. Figures of more complex pore structures are actually found in manymulti-porositypapers. But if it is the case that the free water part is not supposed to be interpreted on the microscopic scale, we are basically back to a magic membrane picture of the structure! Moreover, if the free water is not supposed to be on the microscopic scale, the diffuse layer will always have a negligible volume, and these illustrations don’t provide a mean for calculating the partitioning between “micro” and “macroporosity”.
It seems to me that not specifying the extension of the free water is a way for authors to dodge the question of how it is actually distributed (and, as a consequence, to not state what constitutes the semi-permeable component).
[7] The PHREEQC input files are provided as
supplementary material to Appelo and Wersin (2007). Here I consider the input corresponding
to figure 3c in the article. The free water is specified with
keyword “SOLUTION”.
[8] Keyword
“SURFACE” in the PHREEQC input file for figure 3c in the paper.
[9] Using the identifier “-donnan” for the “SURFACE”
keyword.
[10] We assume a boundary
condition such that the potential is zero in the solution infinitely
far away from any clay component.
[11] Assuming exponential decay, which is only strictly true for a single clay layer of low charge.
[12] For example,
Tournassat and Steefel
(2019) write
(\(f_{DL}\) denotes the volume fraction of the diffuse layer):
In PHREEQC and CrunchClay, the volume of the diffuse layer (\(V_{DL}\) in m3), and hence the \(f_{DL}\) value, can be defined as a multiple of the Debye length in order to capture this effect of ionic strength on \(f_{DL}\): \begin{equation*} V_{DL} = \alpha_{DL}\kappa^{-1}S \tag{22} \end{equation*} \begin{equation*} f_{DL} = V_{DL}/V_{pore} \end{equation*} […] it is obvious that \(f_{DL}\) cannot exceed 1. Equation (22) must then be seen as an approximation, the validity of which may be limited to small variations of ionic strength compared to the conditions at which \(f_{DL}\) is determined experimentally. This can be appreciated by looking at the results obtained with a simple model where: \begin{equation*} \alpha_{DL} = 2\;\mathrm{if}\;4\kappa^{-1} \le V_{pore}/S\;\mathrm{and,} \end{equation*} \begin{equation*} f_{DL} = 1 \;\mathrm{otherwise.} \end{equation*}
[13] Some tools (e.g. PHREEQC) allow to put a maximum size limit on the diffuse layer domain, independent of chemical conditions. This is of course only a way for the code to “work” under all conditions.
[14] As icing on the cake, these estimations of free water in bentonite (15%) and Opalinus clay (50%) appear to be based on the incorrect assumption that “anions” only reside in such compartments. In the present context, this handling is particularly confusing, as a main point with multi-porosity models (I assume?) is to evaluate ion concentrations in other types of compartments.
[16] Donnan equilibrium between microscopic
compartments can be studied in
molecular dynamics simulations, but they require the considered
system to be large enough for the electrostatic potential to reach
zero. The semi-permeable component in such simulations is
implemented by simply imposing constraints on the atoms making up
the clay layer.
We havediscussedvariousaspects of “anionexclusion” on this blog. This concept is often used to justify multi-porosity models of compacted bentonite, by reasoning that the exclusion mechanism makes parts of the pore space inaccessible to anions. But we have seen that this reasoning has no theoretical backing: studies making such assumptions usually turn out to refer to conventional electric double layer theory, described e.g. by the Poisson-Boltzmann equation. In the following, we refer to the notion of compartments inaccessible to anions as complete anion exclusion.
In fact, a single, physically reasonable concept underlies basically all descriptions of anion exclusion in the clay literature: charge separation. Although the required mathematics may differ for different systems — may it be using Donnan’s “classical equations”, or the Poisson-Boltzmann equation — the underlying mechanism is the same. In the following we refer to this type of description as traditional theory or Donnan theory. It is important to recognize that traditional theory is incompatible with complete anion exclusion: the Poisson-Boltzmann equation predicts anions everywhere.
In more recent years, however, a different meaning of the term “anion
exclusion” has sneaked into the literature. This seems to be related
to the dawn of molecular dynamics (MD) simulations of clays. In
particular, the study of Rotenberg et al. (2007) — which I think is the first published MD
simulation of montmorillonite interlayers in contact with an external
compartment — is frequently cited as demonstrating qualitatively
different results as compared with the traditional
models. E.g. Kosakowski and Berner (2013) write
Very often it is assumed that negatively charged ions are strongly hindered to enter the interlayer space (Kosakowski et al., 2008; Rotenberg et al., 2007), although other authors come to different conclusions (Karnland et al., 2007). Note that we favor the former view with our montmorillonite setup.
Although the terms “assumed” and “conclusions” seem misplaced, it
is clear that Kosakowski and Berner (2013) mean that the interlayer space is
essentially anion-free, rather than obeying ordinary Donnan
equilibrium (the approach used in
Karnland et
al. (2007)).
The interlayer space can be seen as an extreme case where the diffuse layer vanishes leaving only the Stern layer of the adjacent basal surfaces. For this reason, the interlayer space is often considered to be completely free of anions (Tournassat and Appelo 2011), although this hypothesis is still controversial (Rotenberg et al. 2007c; Birgersson and Karnland 2009).
Based upon [results from anion diffusion tests], anion-exclusion models have been formulated, which subdivide the water-filled pore space into interlayer, diffuse (or electric) double layer (DDL) and “free” water porosities (Wersin et al. 2004; Tournassat & Appelo 2011; Appelo 2013). In this formulation, anions are considered to reside in the “free” electrically neutral solution and in the DDL in the external (intergranular) pores, whereas the interlayer (intragranular) space is considered devoid of anions. Support for this model has been given by molecular dynamics simulations (Rotenberg et al. 2007), but this issue remains controversial (Birgersson & Karnland 2009)
The term “anion-exclusion” is here fully transformed to refer to complete exclusion, rather than to the traditional theory from which the term was coined. Note that the picture of bentonite given in this and the previous quotations is basically the contemporary mainstream view, which we discussed in a previous blog post. This description has not emerged from considering MD results that are allegedly in contradiction with traditional Donnan equilibrium theory. Rather, it has resulted from misusing the concept of exclusion-volume. The study of Rotenberg et al. (2007) (Rot07, in the following) supports the contemporary mainstream view only to the extent that it is at odds with the predictions of traditional theory. But is it? Let’s take a look at the relevant MD studies.
Rotenberg et al. (2007)
Rot07 is not primarily a study of the anion equilibrium, but considers more generally the transition of species between an external compartment2 and interlayer pores: water, cations (Na and Cs), and anions (Cl). The study only concerns interlayers with two monolayers of water, in the following referred to as a 2WL system. There is of course nothing wrong with exclusively studying the 2WL system, but this study alone cannot be used to support general model assumptions regarding interlayers (which anyway is commonplace, as we saw above). The meaning of the term “interlayer” in modern clay literature is quite confusing, but there is at least full consensus that it includes also states with three monolayers of water (3WL) (we’ll get back to those). Rot07 furthermore consider only a single external concentration, of 0.52 M.
Here is an illustration of the simulated system:
A cell (outlined with dashed lines) containing two montmorillonite
layers (yellow) and six chloride ions (green) is repeated infinitely
in all directions (the cell depth in the direction normal to the
picture is 20.72 Å). While only chloride ions are indicated in this
figure, also cations, water atoms, and montmorillonite atoms are
explicitly accounted for in the simulation.
Note that the study neither varies density (interlayer distance) nor
external concentration (number of chloride ions) — two variables
essential for studying anion equilibrium. I don’t mean this as direct
criticism, but it should be recognized when the study is used to
support assumptions regarding interlayers in other models.
What I do want to criticize, however, is that
Rot07 don’t
actually compare with Donnan theory. Instead, they seem to be under
the impression that traditional theory predicts complete exclusion in
their system. Consider this passage in the introduction
Due to the negative charge of clay layers, anions should be repelled by the external surfaces, and excluded from the interlayers. On the contrary, cations are attracted by the surfaces, and may exchange with the natural interlayer counterions.
Here they associate two different terms with the anions: they are
repelled by the “external surfaces” and excluded from
“interlayers”.
I can only interpret this as meaning that anions are completely
excluded from interlayers, especially as the wording “on the
contrary” is used when describing cations.3
The study comprises both a “plain” MD simulation of the (presumed) equilibrium state, and separate calculations of free energy profiles. In the “plain” MD simulation, anions do not enter the interlayers, and the calculation of the free energy profile gives a barrier of ~9 kT for chloride to enter the interlayer.
These results motivate the authors to conclude that the “thermal fluctuations do not allow anions to overcome the free energy barrier corresponding to their entrance into the interlayer” and that “anions are excluded from the interlayer: the probability for an anion reaching the interface to enter into the interlayer is very small (of the order of e-9 ~ 10-4)”
It is important to keep in mind that the authors are under the
impression that this result and conclusion are in line with the
traditional description of anion
exclusion.3 When summarizing
their findings they write
All the results are in agreement with the common sense on ionic exchange and anion exclusion.
and
The results confirm the generally admitted ideas of ionic exchange and anion exclusion
The problem is that this “common sense” and these “generally
admitted ideas” are based on
misconceptions of traditional theory (I also think one should be
careful with using terms like these in scientific
writing). Consequently, the authors erroneously conclude that their
results confirm, rather than contrast, traditional theory. This is
opposite to how this study is referred to in later publications, as
was exemplified above.
The anion exclusion predicted from Donnan theory for the system in
Rot07 is estimated
as follows. The adopted montmorillonite unit cell
(Na0.75Si8Al3.25Mg0.75O20OH4)
has
structural charge 0.75e, and lateral dimensions 8.97 Å × 5.18
Å. With an interlayer width of 6.1 Å we thus have for the
concentration of interlayer charge
where \(N_A\) is the
Avogadro
constant. Using this value for \(c_{IL}\) in the expression for
internal anion concentration in an ideal
1:1 Donnan system,
This should be the anion interlayer concentration expected from “generally admitted ideas”, and Rot07 should have concluded that their results differ by a factor ~1000 (or more) from traditional theory. This is not to say that the calculations are incorrect (more on that later), but it certainly puts the results in a different light. A discrepancy of this magnitude should reasonably be of interest to investigate further.
Hsiao and Hedström (2015)
Considerably more detailed MD simulations of the 2WL system are
provided by Hsiao and
Hedström (2015) (Hsi15, hereafter). In contrast to
Rot07,
Hsi15 specifically
focus on the anion equilibrium, and they explicitly compare with both
conventional Donnan theory, and the results of
Rot07. In these
simulations, chloride actually populates the interlayer.
Hsi15 also analyze the convergence behavior, by varying system size and simulation time. This analysis makes it clear both that most of the simulations presented in the paper are properly converged, and that the simulation of Rot07 is not. With external concentration 1.67 M, Hsi15 demonstrate that, during intervals of 20 ns, the interlayer concentration fluctuates between basically zero and 0.13 M (converged value: 0.04 M), in a system with similar size as that of Rot07. Given that the total simulation time of the earlier study is 20 ns, and that it also adopts a considerably lower external concentration, its result of zero chloride concentration in the interlayer is no surprise.
The converged interlayer concentrations in
Hsi15 look like
this in the direction normal to the basal surfaces (simulation time:
150 ns, layer size: 8 × 4 unit cells, external concentration:
1.67 M)
Note that the simulation contains two interlayer pores (indicated by the dotted lines; cf. the illustration of the simulated system) and that sodium and chloride populate the same central layer, sandwiched by the two water layers (not shown). The nearly identical chloride profiles are a strong confirmation that the simulation is converged.
The chloride interlayer concentrations evaluated in Hsi15 deviate strongly from the predictions of the ideal Donnan formula. With \(c_{IL}\) = 4.23 M (as reported in the article) and \(c^\mathrm{ext}\) = 1.67 M, eq. 1 gives \(c^\mathrm{int}\) = 0.580 M, while the MD results are in the range 0.033 M — 0.045 M, i.e. more than a factor 10 lower (but not a factor 1000).
Hsi15 also calculate
the free energy profiles along the coordinate connecting the external
compartment and the interlayer, similar to the technique utilized by
Rot07 (as far as I
understand). For the external concentration of 1.67 M they evaluate a
free energy barrier of ~3.84 kT, which corresponds to an
interlayer concentration of 0.036 M, and is in good agreement with the
directly evaluated concentrations.
Note that Hsi15 —
in contrast to Rot07 — conclude significant deviation between the MD results of
the 2WL system and ideal traditional theory. Continuing their
investigation (again, in contrast to
Rot07),
Hsi15 found that the
contribution from ion hydration to the free energy barrier basically
make up for the entire discrepancy with the ideal Donnan formula.
Moreover, even though the ideal Donnan formula strongly overestimates
the actual values obtained from MD, it still shows the correct
dependency on external concentration: when the external
concentration is lowered to 0.55 M, the evaluated free energy barrier
increases to ~5.16 kT, which corresponds to a reduction of the
internal concentration by about a factor of 10. This is in
agreement with Donnan theory, which gives for the expected
reduction (0.55/1.67)2 ≈ 0.11.
From the results of Hsi15 (and Rot07,
for that matter), a relatively clear picture emerges: MD simulated 2WL
systems function as Donnan systems. Anions are not completely
excluded, and the dependency on external concentration is in line with
what we expect from
a varying Donnan potential across the interface between interlayer
and external compartment
(Hsi15 even comment
on observing the space-charge region!).
The simulated 2WL system is, however, strongly non-ideal, as a consequence of the ions not being optimally hydrated. Hsi15 remark that the simulations probably overestimate this energy cost, e.g. because atoms are treated as non-polarizable. This warning should certainly be seriously considered before using the results of MD simulated 2WL systems to motivate multi-porosity in compacted bentonite. But, concerning assumptions of complete anion exclusion in interlayers, another system must obviously also be considered: 3WL.
Hedström and Karnland (2012)
MD simulations of anion equilibrium in the 3WL system are presented in
Hedström and
Karnland (2012) (Hed12, in the following).
Hed12 consider
three different external concentrations, by including either 12, 6, or
4 pairs of excess ions (Cl– + Na+). This study
also varies the way the interlayer charge is distributed, by either
locating unit charges on specific magnesium atoms in the
montmorillonite structure, or by evenly reducing the charge by a minor
amount on all the octahedrally coordinated atoms.
Here are the resulting ion concentration profiles across the
interlayer, for the simulation containing 12 chloride ions, and evenly
distributed interlayer charge (simulation time: 20 ns, layer size:
4 × 4 unit cells)
Chloride mainly resides in the middle of the interlayer also in the 3WL system, but is now separated from sodium, which forms two off-center main layers. The dotted lines indicate the extension of the interlayer.
The main objectives of this study are to simply establish that anions in MD equilibrium simulations do populate interlayers, and to discuss the influence of unavoidable finite-size effects (6 and 12 are, after all, quite far from Avogadro’s number). In doing so, Hed12 demonstrate that the system obeys the principles of Donnan equilibrium, and behaves approximately in accordance with the ideal Donnan formula (eq. 1). The authors acknowledge, however, that full quantitative comparison with Donnan theory would require better convergence of the simulations (the convergence analysis was further developed in Hsi15). If we anyway make such a comparison, it looks like this
#Cl TOT
Layer charge
#Cl IL
\(c^\mathrm{ext}\)
\(c^\mathrm{int}\) (Donnan)
\(c^\mathrm{int}\) (MD)
12
distr.
1.8
1.45
0.62
0.42 (67%)
12
loc.
1.4
1.50
0.66
0.32 (49%)
6
distr.
0.6
0.77
0.20
0.14 (70%)
6
loc.
1.3
0.67
0.15
0.30 (197%)
4
distr.
0.2
0.54
0.10
0.05 (46%)
4
loc.
0.18
0.54
0.10
0.04 (41%)
The first column lists the total number of chloride ions in the simulations, and the second indicates if the layer charge was distributed on all octahedrally coordinated atoms (“distr.”) or localized on specific atoms (“loc.”) The third column lists the average number of chloride ions found in the interlayer in each simulation. \(c^\mathrm{ext}\) denotes the corresponding average molar concentration in the external compartment. The last two columns lists the corresponding average interlayer concentration as evaluated either from the Donnan formula (eq. 1 with \(c_{IL}\) = 2.77 M, and the listed \(c^\mathrm{ext}\)), or from the simulation itself.
The simulated results are indeed within about a factor of 2 from the predictions of ideal Donnan theory, but they also show a certain variation in systems with the same number of total chloride ions,4 indicating incomplete convergence (compare with the fully converged result of Hsi15). It is also clear from the analysis in Hed12 and Hsi15 that the simulations with the highest number of chloride ions (12) are closer to being fully converged.5 Let’s therefore use the result of those simulations to compare with experimental data.
Comparison with experiments
In an earlier blog post, we looked at the available experimental data on chloride equilibrium concentrations in Na-dominated bentonite. Adding the high concentration chloride equilibrium results from Hed12 and Hsi15 to this data (in terms of \(c^\mathrm{int}/c^\mathrm{ext}\)), gives the following picture6 (the 3WL system corresponds to pure montmorillonite of density ~1300 kg/m3, and the 2WL system corresponds to ~1600 kg/m3, as also verified experimentally).
The x-axis shows montmorillonite effective dry density, and applied external concentrations for each data series are color coded, but also listed in the legend. Note that this plot contains mainly all available information for drawing conclusions regarding anion exclusion in interlayers.7 To me, the conclusions that can be drawn are to a large extent opposite to those that have been drawn:
The amount chloride in the simulated 3WL system corresponds roughly to measured values. Consequently, MD simulations do not support models that completely exclude anions from interlayers.
The 3WL results instead suggest that interlayers contain the main contribution of chloride. Interlayers must consequently be handled no matter how many additional pore structures a model contains.
For systems corresponding to 2WL interlayers, there is a choice: Either,
assume that the discrepancy between simulations and measurements indicates the existence of an additional pore structure, where the majority of chloride resides, or
assume that presently available MD simulations of 2WL systems overestimate “anion” exclusion.8
Tournassat et al. (2016) (Tou16, in the following) present more MD simulations of interlayer pores in contact with an external compartment, with a fixed amount of excess ions, at three different interlayer distances: 2WL (external concentration ~0.5 M), 3WL (~0.4 M), and 5WL (~0.3 M).
In the 2WL simulations, no anions enter the interlayers. Tou16 do not reflect on the possibility that 2WL simulations may overestimate exclusion, as suggested by Hsi15,9 but instead use this result to argue that anions are basically completely excluded from 2WL interlayers. They even imply that the result of Rot07 is more adequate than that of Hsi15
In the case of the 2WL hydrate, no Cl– ion entered the interlayer space during the course of the simulation, in agreement with the modeling results of Rotenberg et al. (2007b), but in disagreement with those of Hsiao and Hedström (2015).
But, as discussed, there is no real “disagreement” between the
results of Hsi15 and
Rot07. To refute
the conclusions of Hsi15, Tou16 are
required to demonstrate well converged results, and analyze what is
supposedly wrong with the simulations of
Hsi15. It is,
furthermore, glaringly obvious that most of the anion equilibrium
results in Tou16
are not converged.
Regarding convergence, the only “analysis” provided is the following
passage
The simulations were carried out at the same temperature (350 K) as the simulations of Hsiao and Hedström (2015) and with similar simulation times (50 ns vs. 100-200 ns) and volumes (27 × 104 Å3vs. 15 × 104 Å3), thus ensuring roughly equally reliable output statistics. The fact that Cl– ions did not enter the interlayer space cannot, therefore, be attributed to a lack of convergence in the present simulation, as Hsiao and Hedström have postulated to explain the difference between their results and those of Rotenberg et al. (2007b).
I mean that this is not a suitable procedure in a scientific
publication — the authors should of course demonstrate convergence of
the simulations actually performed! (Especially after
Hsi15 have provided
methods for such an analysis.10)
Anyhow, Tou16 completely miss that Hsi15 demonstrate convergence in simulations with external concentration 1.67 M; for the system relevant here (0.55 M), Hsi15 explicitly write that the same level of convergence requires a 10-fold increase of the simulation time (because the interlayer concentration decreases approximately by a factor of 10, as predicted by — Donnan theory). Thus, the simulation time of Tou16 (53 ns) should be compared with 2000 ns, i.e. it is only a few percent of the time required for proper convergence.
Further confirmation that the simulations in
Tou16 are not
converged is given by the data for the systems where chloride
has entered the interlayers. The ion concentration profiles for
the 3WL simulation look like this
The extension of the interlayers is indicated by the dotted lines. Each interlayer was given slightly different (average) surface charge density, which is denoted in the figure. One of the conspicuous features of this plot is the huge difference in chloride content between different interlayers: the concentration in the mid-pore (0.035 M) is more than three times that in left pore (0.010 M). This clearly demonstrates that the simulation is not converged (cf. the converged chloride result of Hsi15). Note further that the larger amount of chloride is located in the interlayer with the highest surface charge, and the least amount is located in the interlayer with the smallest surface charge.11 I think it is a bit embarrassing for Clays and Clay Minerals to have used this plot for the cover page.
As the simulation times (53 ns vs. 40 ns), as well as the external concentrations (~0.5 M vs. ~0.4 M), are similar in the 2WL and and 3WL simulations, it follows from the fact that the 3WL system is not converged, that neither is the 2WL system. In fact, the 2WL system is much less converged, given the considerably lower expected interlayer concentration. This conclusion is fully in line with the above consideration of convergence times in Hsi15.
For chloride in the 3WL (and 5WL) system, Tou16 conclude that “reasonable quantitative agreement was found” between MD and traditional theory, without the slightest mentioning of what that implies.12 I find this even more troublesome than the lack of convergence. If the authors mean that MD simulations reveal the true nature of anion equilibrium (as they do when discussing 2WL), they here pull the rug out from under the entire mainstream bentonite view! With the 3WL system containing a main contribution, interlayers can of course not be modeled as anion-free, as we discussed above. Yet, not a word is said about this in Tou16.
In this blog post I have tried to show that available MD simulations do not, in any reasonable sense, support the assumption that anions are completely excluded from interlayers. Frankly, I see this way of referencing MD studies mainly as an “afterthought”, in attempts to justify the misuse of the exclusion-volume concept. In this light, I am not surprised that Hed12 and Hsi15 have not gained reasonable attention, while Tou16 nowadays can be found referenced to support claims that anions do not have access to “interlayers”.13
Footnotes
[1] I should definitely discuss the “Stern layer” in a future blog post. Update (250113): Stern layers are discussed here.
[2] The view of bentonite (“clay”) in Rotenberg et al. (2007) is strongly rooted in a “stack” concept. What I refer to as an “external compartment” in their simulation, they actually conceive of as a part of the bentonite structure, calling it a “micropore”.
[3] That
Rotenberg et
al. (2007) expresses this view of anion exclusion puzzles me
somewhat, since several of the same authors published a study just a
few years later where Donnan theory was explored in similar systems:
Jardat et al. (2009).
[4] Since the number of chloride ions found in the
interlayer is not correlated with how layer charge is distributed,
we can conclude that the latter parameter is not important for the
process.
[5] The small difference in the two
simulations with 4 chloride ions is thus a coincidence.
[6] I am in the process of
assessing the experimental data, and hope to be able to better
sort out which of these data series are more relevant. So far I have
only looked at — and discarded —
the
study by Muurinen et al. (1988). This study is therefore removed
from the plot.
[7] There are of course severalotherresults that indirectly demonstrate the presence of anions in interlayers. Anyway, I think that the bentonite research community, by now, should have managed to produce better concentration data than this (both simulated and measured).
[8] As the cation (sodium) may give a major contribution to the hydration energy barrier (this is not resolved in Hsiao and Hedström (2015)), it may be inappropriate to refer to this part as “anion” exclusion (remember that it is salt that is excluded from bentonite). It may be noted that sodium actually appear to have a hydration barrier in e.g. the Na/Cs exchange process, which has been explored both experimentally and in MD simulations.
[9] Tournassat et al. (2016) even refer to Hsiao and Hedström (2015) as presenting a “hypothesis” that “differences in solvation energy play an important role in inhibiting the entry of Cl– in the interlayer space”, rather than addressing their expressed concern that the hydration energy cost may be overestimated.
[11] As the interlayers have different surface charge, they are not expected to have identical chloride content. But the chloride content should reasonably decrease with increasing surface charge, and the difference between interlayers should be relatively small.
[12] Here we have to disregard that the “agreement” is not quantitative. It is not even qualitative: the highest chloride content was recorded in the interlayer pore with highest charge, in both the 3WL and the 5WL system.
where \(\phi\) is the porosity of the sample, \(D_c\) is the macroscopic
pore diffusivity of the presumed interlayer domain, and \(\Xi\) is the
ion equilibrium coefficient. \(\Xi\) quantifies the ratio between
internal and external concentrations of the ion under consideration,
when the two compartments are in equilibrium.
where \(\epsilon_\mathrm{eff}\) is the porosity of a presumed bulk water
domain where anions are assumed to reside exclusively, and \(D_p\) is
the corresponding pore diffusivity of this bulk water domain.
We have
discussed earlier
how the homogeneous mixture and the effective porosity models can be
equally well fitted to a specific set of anion through-diffusion
data. The parameter “translation” is simply
\(\phi\cdot \Xi \leftrightarrow \epsilon_\mathrm{eff}\) and
\(D_c \leftrightarrow D_p\). It may appear from this equivalency that
diffusion data alone cannot be used to discriminate between the two
models.
But note that the interpretation of how \(D_e\) varies with background
concentration is very different in the two models.
In the homogeneous mixture model, \(D_c\) is not expected to vary with background concentration to any greater extent, because the diffusing domain remains essentially the same. \(D_e\) varies in this model primarily because \(\Xi\) varies with background concentration, as a consequence of an altered Donnan potential.
In the effective porosity model, \(D_p\) is expected to vary, because the volume of the bulk water domain, and hence the entire domain configuration (the “microstructure”), is postulated to vary with background concentration. \(D_e\) thus varies in this model both because \(D_p\) and \(\epsilon_\mathrm{eff}\) varies.
A simple way of taking into account a varying domain configuration (as in the effective porosity model) is to assume that \(D_p\) is proportional to \(\epsilon_\mathrm{eff}\) raised to some power \(n – 1\), where \(n > 1\). Eq. 2 can then be written
where \(D_0\) is the tracer diffusivity in pure bulk water. Eq. 3 is in the bentonite literature often referred to as “Archie’s law”, in analogy with a similar evaluation in more conventional porous systems. Note that with \(D_0\) appearing in eq. 3, this expression has the correct asymptotic behavior: in the limit of unit porosity, the effective diffusivity reduces to that of a pure bulk water domain.
Eq. 3 shows that \(D_e\) in the effective porosity model is expected to depend non-linearly on background concentration for constant sample density. In contrast, since \(D_c\) is not expected to vary significantly with background concentration, we expect a linear dependence of \(D_e\) in the homogeneous mixture model. Keeping in mind the parameter “translation” \(\phi\cdot\Xi \leftrightarrow \epsilon_\mathrm{eff}\), the prediction of the homogeneous mixture model (eq. 1) can be expressed1
We have thus managed to establish a testable difference between the effective porosity and the homogeneous mixture model (eqs. 3 and 4). This is is great! Making this comparison gives us a chance to increase our process understanding.
Comparison with experiment
Van Loon et al. (2007)
It turns out that the chloride diffusion measurements performed by Van Loon et al. (2007) are accurate enough to resolve whether \(D_e\) depends on “\(\epsilon_\mathrm{eff}\)” according to eqs. 3 or 4. As will be seen below, this data shows that \(D_e\) varies in accordance with the homogeneous mixture model (eq. 4). But, since Van Loon et al. (2007) themselves conclude that \(D_e\) obeys Archie’s law, and hence complies with the effective porosity model, it may be appropriate to begin with some background information.
Van Loon et al. (2007) report three different series of diffusion tests, performed on bentonite samples of density 1300, 1600, and 1900 kg/m3, respectively. For each density, tests were performed at five different NaCl background concentrations: 0.01 M, 0.05 M, 0.1 M, 0.4 M, and 1.0 M. The tests were evaluated by fitting the effective porosity model, giving the effective diffusion coefficient \(D_e\) and corresponding “effective porosity” \(\epsilon_\mathrm{eff}\) (it is worth repeating that the latter parameter equally well can be interpreted in terms of an ion equilibrium coefficient).
Van Loon et al. (2007) conclude that their data complies with eq. 3, with \(n = 1.9\), and provide a figure very similar to this one
Here are compared evaluated values of effective diffusivity and “effective porosity” in various tests. The test series conducted by Van Loon et al. (2007) themselves are labeled with the corresponding sample density, and the literature data is from García-Gutiérrez et al. (2006)2 (“Garcia 2006”) and the PhD thesis of A. Muurinen (“Muurinen 1994”). Also plotted is Archie’s law with \(n\) =1.9. The resemblance between data and model may seem convincing, but let’s take a further look.
Rather than lumping together a whole bunch of data sets, let’s focus on the three test series from Van Loon et al. (2007) themselves, as these have been conducted with constant density, while only varying background concentration. This data is thus ideal for the comparison we are interested in (we’ll get back to commenting on the other studies).
It may also be noted that the published plot contains more data points (for these specific test series) than are reported in the rest of the article. Let’s therefore instead plot only the tabulated data.3 The result looks like this
Here we have also added the predictions from the homogeneous mixture model (eq. 4), where \(D_c\) has been fitted to each series of constant density.
The impression of this plot is quite different from the previous one: it should be clear that the data of Van Loon et al. (2007) agrees fairly well with the homogeneous mixture model, rather than obeying Archie’s law. Consequently, in contrast to what is stated in it, this study refutes the effective porosity model.
The way the data is plotted in the article is reminiscent of Simpson’s paradox: mixing different types of dependencies of \(D_e\) gives the illusion of a model dependence that really isn’t there. Reasonably, this incorrect inference is reinforced by using a log-log diagram (I have warned about log-log plots earlier). With linear axes, the plots give the following impression
This and the previous figure show that \(D_e\) depends approximately linearly on “\(\epsilon_\mathrm{eff}\)”, with a slope dependent on sample density. With this insight, we may go back and comment on the other data points in the original diagram.
García-Gutiérrez et al. (2006) and Muurinen et al. (1988)
The tests by García-Gutiérrez et al. (2006) don’t vary the background concentration (it is not fully clear what the background concentration even is4), and each data point corresponds to a different density. This data therefore does not provide a test for discriminating between the models here discussed.
I have had no access to Muurinen (1994), but by examining the data, it is clear that it originates from Muurinen et al. (1988), which was assessed in detail in a previous blog post. This study provides two estimations of “\(\epsilon_\mathrm{eff}\)”, based on either breakthrough time or on the actual measurement of the final state concentration profile. In the above figure is plotted the average of these two estimations.5
One of the test series in Muurinen et al. (1988) considers variation of density while keeping background concentration fixed, and does not provide a test for the models here discussed. The data for the other two test series is re-plotted here, with linear axis scales, and with both estimations for “\(\epsilon_\mathrm{eff}\)”, rather than the average6
As discussed in the assessment of this study, I judge this data to be too uncertain to provide any qualitative support for hypothesis testing. I think this plot confirms this judgment.
Glaus et al. (2010)
The measurements by Van Loon et al. (2007) are enough to convince me that the dependence of \(D_e\) for chloride on background concentration is furtherevidence for that a homogeneous view of compacted bentonite is principally correct. However, after the publication of this study, the same authors (partly) published more data on chloride equilibrium, in pure Na-montmorillonite and “Na-illite”,7 in Glaus et al. (2010).
This data certainly shows a non-linear relation between \(D_e\) and “\(\epsilon_\mathrm{eff}\)” for Na-montmorillonite, and Glaus et al. (2010) continue with an interpretation using “Archie’s law”. Here I write “Archie’s law” with quotation marks, because they managed to fit the expression to data only by also varying the prefactor. The expression called “Archie’s law” in Glaus et al. (2010) is
where \(A\) is now a fitting parameter. With \(A\) deviating from \(D_0\), this expression no longer has the correct asymptotic behavior as expected when interpreting \(\epsilon_\mathrm{eff}\) as quantifying a bulk water domain (see eq. 3). Nevertheless, Glaus et al. (2010) fit this expression to their measurements, and the results look like this (with linear axes)
Here is also plotted the prediction of the homogeneous mixture model
(eq. 4). For the montmorillonite data, the dependence is
clearly non-linear, while for the “Na-illite” I would say that the
jury is still out.
Although the data for montmorillonite in
Glaus et al. (2010)
is
non-linear, there are several strong arguments for why this is not an
indication that the effective porosity model is correct:
Remember that this result is not a confirmation of the measurements in Van Loon et al. (2007). As demonstrated above, those measurements complies with the homogeneous mixture model. But even if accepting the conclusion made in that publication (that Archie’s law is valid), the Glaus et al. (2010) results do not obey Archie’s law (but “Archie’s law”).
The four data points correspond to background concentrations of 0.1 M, 0.5 M, 1.0 M, and 2.0 M. If “\(\epsilon_\mathrm{eff}\)” represented the volume of a bulk water phase, it is expected that this value should level off, e.g. as the Debye screening length becomes small (Van Loon et al. (2007) argue for this). Here “\(\epsilon_\mathrm{eff}\)” is seen to grow significantly, also in the transition between 1.0 M and 2.0 M background concentration.
These are Na-montmorillonite samples of dry density 1.9 g/cm3. With an “effective porosity” of 0.067 (the 2.0 M value), we have to accept more than 20% “free water” in these very dense systems! This is not even accepted by otherproponents of bulk water in compacted bentonite.
Furthermore, these tests were performed with a background of \(\mathrm{NaClO_4}\), in contrast to Van Loon et al. (2007), who used chloride also for the background. The only chloride around is thus at trace level, and I put my bet on that the observed non-linearity stems from sorption of chloride on some system component.
Insight from closed-cell tests
Note that the issue whether or not \(D_e\) varies linearly with
“\(\epsilon_\mathrm{eff}\)” at constant sample density is equivalent
to whether or not \(D_p\) (or \(D_c\)) depends on background
concentration. This is similar to how presumed concentration
dependencies of the pore diffusivity for simple cations
(“apparent”
diffusivities) have been used to argue for multi-porosity in compacted
bentonite. For cations,
a closer look shows that no such dependency is found in the
literature.
For anions, it is a bit frustrating that the literature data is not
accurate or relevant enough to fully settle this issue (the data of
Van Loon et al. (2007)
is, in my opinion, the best available).
However, to discard the conceptual view underlying the effective porosity model, we can simply use results from closed-cell diffusion studies. In Na-montmorillonite equilibrated with deionized water, Kozaki et al. (1998) measured a chloride diffusivity of \(1.8\cdot 10^{-11}\) m2/s at dry density 1.8 g/cm3.8 If the effective porosity hypothesis was true, we’d expect a minimal value for the diffusion coefficient9 in this system, since \(\epsilon_\mathrm{eff}\) approaches zero in the limit of vanishing ionic strength. Instead, this value is comparable to what we can evaluate from e.g. Glaus et al. (2010) at 1.9 cm3/g, and 2.0 M background electrolyte: \(D_e/\epsilon_\mathrm{eff} = 7.2\cdot 10^{-13}/0.067\) m2/s = \(1.1\cdot 10^{-11}\) m2/s.
That chloride diffuses just fine in dense montmorillonite equilibrated with pure water is really the only argument needed to debunk the effective porosity hypothesis.
Footnotes
[1] Note that \(\epsilon_\mathrm{eff}\) is not a parameter in the homogeneous mixture model, so eq. 4 looks a bit odd. But it expresses \(D_e\) if \(\phi\cdot \Xi\) is interpreted as an effective porosity.
[3] This choice is not critical for the conclusions made in this blog post, but it seems appropriate to only include the data points that are fully described and reported in the article.
[4] García-Gutiérrez et al. (2004) (which is the study compiled in García-Gutiérrez et al. (2006)) state that the samples were saturated with deionized water, and that the electric conductivity in the external solution were in the range 1 — 3 mS/cm.
[5] The data point labeled with a “?” seems to have been obtained by making this average on the numbers 0.5 and 0.08, rather than the correctly reported values 0.05 and 0.08 (for the test at nominal density 1.8 g/cm3 and background concentration 1.0 M).
[6] Admittedly, also the data we have plotted from the original tests in Van Loon et al. (2007) represents averages of several estimations of “\(\epsilon_\mathrm{eff}\)”. We will get back to the quality of this data in a future blog post when assessing this study in detail, but it is quite clear that the estimation based on the direct measurement of stable chloride is the more robust (it is independent of transport aspects). Using these values for “\(\epsilon_\mathrm{eff}\)”, the corresponding plot looks like this
[7] To my mind, it is a misnomer to describe something as illite in sodium form. Although “illite” seems to be a bit vaguely defined, it is clear that it is supposed to only contain potassium as counter-ion (and that these ions are non-exchangeable; the basal spacing is \(\sim\)10 Å independent of water conditions). The material used in Glaus et al. (2010) (and severalotherstudies) has a stated cation exchange capacity of 0.22 eq/kg, which in a sense is comparable to the montmorillonite material (a factor 1/4). Shouldn’t it be more appropriate to call this material e.g. “mixed-layer”?
[8] This value is the average from two tests performed at 25 °C. The data from this study is better compiled in Kozaki et al. (2001).
[9] Here we refer of course to the empirically defined diffusion coefficient, which I have named \(D_\mathrm{macr.}\) in earlier posts. This quantity is model independent, but it is clear that it should be be associated with the pore diffusivities in the two models here discussed (i.e. with \(D_c\) in the homogeneous mixture model, and with \(D_p\) in the effective porosity model).
On the surface, “Ionendiffusion in Hochverdichtetem Bentonit”1 by G. Kahr, R. Hasenpatt, and M. Müller-Vonmoos, published by NAGRA in March 1985, looks like an ordinary mundane 37-page technical report. But it contains experimental results that could have completely changed the history of model development for compacted clay.
Test principles
The tests were conducted in a quite original manner. By compacting
granules or powder, the investigators obtained samples that
schematically look like this
The bentonite material — which was either Na-dominated “MX-80”, or Ca-dominated “Montigel” — was conditioned to a specific water-to-solid mass ratio \(w\). At one of the faces, the bentonite was mixed with a salt (in solid form) to form a thin source for diffusing ions. This is essentially the full test set-up! Diffusion begins as soon as the samples are prepared, and a test was terminated after some prescribed amount of time, depending on diffusing ion and water content. At termination, the samples were sectioned and analyzed. In this way, the investigators obtained final state ion distributions, which in turn were related to the initial states by a model, giving the diffusion coefficients of interest.
Note that the experiments were conducted without exposing samples to a liquid (external) solution; the samples were “unsaturated” to various degree, and the diffusing ions dissolve within the bentonite. The samples were not even confined in a test cell, but “free-standing”, and consequently not under pressure. They were, however, stored in closed vessels during the course of the tests, to avoid changes in water content.
With this test principle a huge set of diffusion tests were
performed, with systematic variation of the following variables:
Bentonite material (“MX-80” or “Montigel”)
Water-to-solid mass ratio (7% — 33%)
Dry density (1.3 g/m3 — 2.1 g/m3 )
Diffusing salt (SrCl2, SrI2, CsCl, CsI, UO2(NO3)2, Th(NO3)4, KCl, KI, KNO3, K2SO4, K2CO3, KF)
Distribution of water in the samples
From e.g. X-ray diffraction (XRD) we know that bentonite water at low water content is distributed in distinct, sub-nm thin films. For simplicity we will refer to all water in the samples as interlayer water, although some of it, reasonably, forms interfaces with air. The relevant point is that the samples contain no bulk water phase, but only interfacial (interlayer) water.
I argueextensively on this blog for that interlayer water is the only relevant water phase also in saturated samples under pressure. In the present case, however, it is easier to prove that this is the case, as the samples are merely pressed bentonite powder at a certain water content; the bentonite water is not pressurized, the samples are not exposed to liquid bulk water, nor are they in equilibrium with liquid bulk water. Since the water in the samples obviously is mobile — as vapor, but most reasonably also in interconnected interlayers — it is a thermodynamic consequence that it distributes as to minimize the chemical potential.
There is a ton of literature on how the montmorillonite basal spacing
varies with water content. Here, we use the neat result from
Holmboe et al. (2012)
that the average interlayer distance varies basically
linearly2 with water content, like this
XRD-studies also show that bentonite water is distributed in rather distinct hydration states, corresponding to 0, 1, 2, or 3 monolayers of water.3 We label these states 0WL, 1WL, 2WL, and 3WL, respectively. In the figure is indicated the approximate basal distances for pure 1WL (12.4 Å), 2WL (15.7 Å), and 3WL (19.0 Å), which correspond roughly to water-to-solid mass ratios of 0.1, 0.2, and 0.3, respectively.
From the above plot, we estimate roughly that the driest samples in
Kahr et al. (1985)
(\(w \sim 0.1\)) are in pure 1WL states, then transitions to a mixture
of 1WL and 2WL states (\(w\sim 0.1 – 0.2\)), to pure 2WL states
(\(w \sim 0.2\)), to a mixture of 2WL and 3WL states
(\(w\sim 0.2 – 0.3\)), and finally to pure 3WL states (\(w\sim 0.3\)).
Results
With the knowledge of how water is distributed in the samples, let’s
take a look at the results of
Kahr et al. (1985).
Mobility of interlayer cations confirmed
The most remarkable results are of qualitative character. It is, for
instance, demonstrated that several cations diffuse far into the
samples. Since the samples only contain interlayer water, this is a
direct proof of ion mobility in the interlayers!
Also, cations are demonstrated to be mobile even when the water
content is as low as 7 or 10 %! As such samples are dominated by 1WL
states, this is consequently evidence for ion mobility in 1WL states.
A more quantitative assessment furthermore shows that the cation diffusivities varies with water content in an almost step-wise manner, corresponding neatly to the transitions between various hydration states. Here is the data for potassium and strontium
This behavior further confirms that the ions diffuse in interlayers,
with an increasing diffusivity as the interlayers widen.
It should also be noted that the evaluated values of the diffusivities
are comparable to — or even larger4 — than
corresponding results from saturated, pressurized tests.
This strongly suggests that interlayer diffusivity dominates also in
the latter types of tests, which also has been
confirmedin more recent years. The
larger implication is that interlayer diffusion is the only relevant
type of diffusion in general in compacted bentonite.
Anions enter interlayers (and are mobile)
The results also clearly demonstrate that anions (iodide) diffuse in systems with water-to-solid mass ratio as low as 7%! With no other water around, this demonstrates that anions diffuse in — and consequently have access to — interlayers. This finding is strongly confirmed by comparing the \(w\)-dependence of diffusivity for anions and cations. Here is plotted the data for iodide and potassium (with the potassium diffusivity indicated on the right y-axis)
The iodide mobility increases as the system transitions from 1WL to 2WL, in a very similar way as for potassium (and strontium). If this is not a proof that the anion diffuse in the same domain as the cation I don’t know what is! Also for iodide the value of the diffusivity is comparable to what is evaluated in water saturated systems under pressure, which implies that interlayer diffusivity dominates generally in compacted bentonite, also for anions.
Dependence of diffusivity on water content and density
A conclusion made in
Kahr et al. (1985),
that I am not sure I fully agree with, is that diffusivity mainly
depends on water content rather than density. As seen in the diagrams
above, the spread in diffusivity is quite substantial for a given
value of \(w\). There is actually some systematic variation here: for
constant \(w\), diffusivity tend to increase with dry density.
Although using unsaturated samples introduces additional variation, the present study provides a convenient procedure to study diffusion in systems with very low water content. A more conventional set-up in this density limit has to deal with enormous pressures (on the order of 100 MPa).
Interlayer chemistry
An additional result is not acknowledged in the report, but is a direct consequence of the observations: the tests demonstrate that interlayers are chemically active. The initially solid salt evidently dissolves before being able to diffuse. Since these samples are not even close to containing a bulk water phase (as discussed above), the dissolution process must occur in an interlayer. More precisely, the salt must dissolve in interface water between the salt mineral and individual montmorillonite layers, as illustrated here
This study seems to have made no impact at all
In the beginning of 1985, the research community devoted to radioactive waste barriers seems to have been on its way to correctly identify diffusion in interlayers as the main transport mechanism, and to recognize how ion diffusion in bentonite is influenced by equilibrium with external solutions.
Already in 1981,
Torstenfelt et al. (1981)
concluded that the
traditional diffusion-sorption model is not valid,
for e.g. diffusion of Sr and Cs, in compacted bentonite. They also
noted, seemingly without realizing the full importance, that these
ions diffused even in unsaturated samples with as low water-to-solid
mass ratio as 10%.
A significant diffusion was observed for Sr in dry clay, although slower than for water saturated clay, Figure 4, while Cs was almost immobile in the dry clay.
A year later also
Eriksen and Jacobsson (1982)
concluded that the traditional diffusion model is not valid. They
furthermore pointed out the subtleties involved when interpreting
through-diffusion experiments, due to ion equilibrium effects
One difficulty in correlating the diffusivities obtained from profile analysis to the diffusivities calculated from steady state transport data is the lack of knowledge of the tracer concentration at the solution-bentonite interface. This concentration is generally higher for sorbing species like positive ions (counterions to the bentonite) and lower for negative ions (coions to the bentonite) as shown schematically in figure 11. The equilibrium concentration of any ion in the bentonite and solution respectively is a function of the ionic charge, the ionic strength of the solution and the overall exchanger composition and thereby not readily calculated
By regarding the clay-gel as a concentrated electrolytic system Marinsky has calculated (30) distribution coefficients for Sr2+ and Cs+ ions in good agreement with experimentally determined Kd-values. The low anionic exchange capacity and hence the low anion concentration in the pore solution caused by Donnan exclusion also explain the low concentrations of anionic tracers within the clay-gel
[…]
For simple cations the ion-exchange process is dominating and there is, as also pointed out by Marinsky (30), no need to suppose that the counterions are immobilized. It ought to be emphasized that for the compacted bentonite used in the diffusion experiments discussed in this report the water content corresponds roughly to 2-4 water molecule layers (31). There is therefore really no “free water” and the measured diffusivity \(\bar{D}\) can be regarded as corresponding approximately to the diffusivity within the adsorbed phase […]
Furthermore, also
Soudek et al. (1984)
had discarded the traditional diffusion-sorption model, identified the
exchangeable cations as giving a dominating contribution to mass
transfer, and used Donnan equilibrium calculations to account for the
suppressed internal chloride concentration.
In light of this state of the research front, the contribution of Kahr et al. (1985) cannot be described as anything but optimal. In contrast to basically all earlier studies, this work provides systematic variation of several variables (most notably, the water-to-solid ratio). As a consequence, the results provide a profound confirmation of the view described by Eriksen and Jacobsson (1984) above, i.e. that interlayer pores essentially govern all physico-chemical behavior in compacted bentonite. A similar description was later given by Bucher and Müller-Vonmoos (1989) (though I don’t agree with all the detailed statements here)
There is no free pore water in highly compacted bentonite. The water in the interlayer space of montmorillonite has properties that are quite different from those of free pore water; this explains the extremely high swelling pressures that are generated. The water molecules in the interlayer space are less mobile than their free counterparts, and their dielectric constant is lower. The water and the exchangeable cations in the interlayer space can be compared to a concentrated salt solution. The sodium content of the interlayer water, at a water content of 25%, corresponds approximately to a 3-n salt solution, or six times the concentration in natural seawater. This more or less ordered water is fundamentally different from that which engineers usually take into account; in the latter case, pore water in a saturated soil is considered as a freely flowing fluid. References to the porosity in highly compacted bentonite are therefore misleading. Highly compacted bentonite is an unfamiliar material to the engineer.
Given this state of the research field in the mid-80s, I find it
remarkable that history took a different turn. It appears as the
results of
Kahr et al. (1985)
made no impact at all (it may be noticed that they themselves analyzed
the results in terms of the traditional diffusion-sorption
model). And rather than that researchers began identifying that
transport in interlayers is the only relevant contribution, the
so-called surface diffusion model gained popularity (it was already promoted by
e.g.
Soudek et al. (1984)
and
Neretnieks and Rasmuson (1983)). Although this
model emphasizes mobility of the exchangeable cations, it is still
centered around the idea that compacted bentonite contains bulk
water.5 Most
modern bentonite models
suffer from similar flaws: they are formulated in terms of bulk water,
while many effects related to interlayers are treated as irrelevant or
optional.
For the case of anion diffusion the historical evolution is maybe even more disheartening. In 1985 the notions of “effective” or “anion-accessible” porosities seem to not have been that widely spread, and here was clear-cut evidence of anions occupying interlayer pores. But just a few years later the idea began to grow that the pore space in compacted bentonite should be divided into regions which are either accessible or inaccessible to anions. As far as I am aware, the first use of the term “effective porosity” in this context was by Muurinen et al. (1988), who, ironically, seem to have misinterpreted the Donnan equilibrium approach presented by Soudek et al. (1984). To this day, this flawed concept is central in many descriptions of compacted clay.
Footnotes
[1] “Ion
diffusion in highly compacted bentonite”
[2] Incidentally, the slope of this line corresponds to a water “density” of 1.0 g/cm3.
[3] This is the region of swelling often
referred to as
“crystalline”.
[4] I’m not sure the evaluation in Kahr et al. (1985) is fully correct. They use the solution to the diffusion equation for an impulse source (a Gaussian), but, to my mind, the source is rather one of constant concentration (set by the solubility of the salt). Unless I have misunderstood, the mathematical expression to be fitted to data should then be an erfc-function, rather than a Gaussian. Although this modification would change the numerical values of the evaluated diffusion coefficients somewhat, it does not at all influence the qualitative insights provided by the study.
[5] I have discussed the surface diffusion model in some detail in previousblogposts.
At the atomic level, montmorillonite is built up of so-called TOT-layers: covalently bonded sheets of aluminum (“O”) and silica (“T”) oxide (including the right amount of impurities/defects). In my mind, such TOT-layers make up the fundamental particles of a bentonite sample. Reasonably, since montmorillonite TOT-layers vary extensively in size, and since a single cubic centimeter of bentonite contains about ten million billions (\(10^{16}\)), they are generally configured in some crazily complicated manner.
Stack descriptions in the literature
But the idea that the single TOT-layer is the fundamental building
block of bentonite is not shared with many of today’s bentonite
researchers. Instead, you find descriptions like e.g. this one, from
Bacle et al. (2016)
Clay mineral particles consist of stacks of parallel
negatively-charged layers separated by interlayer
nanopores. Consequently, compacted smectite contains two major
classes of pores: interlayer nanopores (located inside the
particles) and larger mesopores (located between the particles).
In compacted rocks, montmorillonite (Mt) forms aggregates
(particles) with 5–20 TOT layers (Segad et al., 2010). A typical
radial size of these particles is of the order of 0.01 to 1
\(\mathrm{\mu m}\). The pore space between Mt particles is referred to
as interparticle porosity. Depending on the degree of compaction,
the interparticle porosity contributes 10 to 30% of the total water
accessible pore space in Mt (Holmboe et al., 2012; Kozaki et al.,
2001).
Such statements show that researchers have something more complex in mind than individual TOT-layers when speaking of “particles”: they are some sort of assemblages of TOT-layers. The quotation of Bacle et al. (2016), using both the terms “stacks” and “particles”, even hints at an idea of a hierarchy of fundamental structures. Such a hierarchy is expressed explicitly in e.g. Navarro et al. (2017), who provide a figure with the caption “Schematic particle arrangement in highly compacted Na-bentonite” that looks similar to this one:
Here it is clear that they differ between “aggregates” (which I’m
not sure is the same thing as “particles”), “stacks”, and
individual TOT-layers (which I assume are represented by the
line-shaped objects). In the following, however, we will use the term
“stack” to refer to any kind of suggested fundamental structure
built up from individual TOT-layers.
The one-sentence version of this blog post is:
Stacks make no sense as fundamental building blocks in models of water saturated, compacted bentonite.
The easiest argument against stacks is, in my mind, to simply work out
the geometrical consequences. But before doing that we will examine
some of the references given to support statements about stacks in
compacted systems. Often, no references are given at all, but when
they are, they usually turn out to be largely irrelevant for the
system under study, or even to support an opposite view.
Inadequate referencing
As an example (of many) of inadequate referencing, we
use the statement above from Churakov et al. (2014) as
a starting point. I think this is a “good” statement, in the sense
that it makes rather precise claims about how compacted bentonite is
supposed to be structured, and provides references for some key
statements, which makes it easier to criticize.
Clay is normally not a homogeneous lamellar material. It might be
better described as a disordered structure of stacks of platelets,
sometimes called tactoids — a tactoid typically consists of 5-20
platelets.19-21
Here the terminology is quite different from the previous quotations: TOT-layers are called “platelets”, and “particles” are called “tactoids”. Still, they use the phrase “stacks of platelets”, so I think we can continue with using “stack” as a sort of common term for what is being discussed.1 We may also note that here is used the word “clay”, rather than “montmorillonite” (as does Bacle et al. (2016)), but it is clear from the context of the article that it really is montmorillonite/bentonite that is discussed.
Anyhow, Segad et al. (2010) do not give much direct information on the claim we investigate, but provide three new references. Two2 of these — Banin (1967) and Shalkevich et al. (2007) — are actually studies on montmorillonite suspensions, i.e. as far away as you can get from compacted bentonite in terms of density; the solid mass fraction in these studies is in the range 1 – 4%.
The average distance between individual TOT-layers in this density limit is comparable with, or even larger than, their typical lateral extension (~100 nm). Therefore, much of the behavior of low density montmorillonite depends critically on details of the interaction between layer edges and various other components, and systems in this density limit behave very differently depending on e.g. ionic strength, cation population, preparation protocol, temperature, time, etc. This complex behavior is also connected with the fact that pure Ca-montmorillonite does not form a sol, while the presence of as little as 10 – 20% sodium makes the system sol forming. The behaviors and structures of montmorillonite suspensions, however, say very little about how the TOT-layers are organized in compacted bentonite.
We have thus propagated from a statement in Churakov et al. (2014), and a similar one in Segad et al. (2010), that montmorillonite in general, in “compacted rocks” forms aggregates of 5 – 20 TOT-layers, to studies which essentially concern different types of materials. Moreover, the actual value of “5 – 20 TOT layers” comes from Banin (1967), who writes
Evidence has accumulated showing that when montmorillonite is
adsorbed with Ca, stable tactoids, containing 5 to 20 parallel
plates, are formed (1). When the mineral is adsorbed with Na, the
tactoids are not stable, and the single plates are separated from
each other.
This source consequently claims that the single TOT-layers are the fundamental units, i.e. it provides an argument against any stack concept! (It basically states that pure Ca-montmorillonite does not form a sol.) In the same manner, even though Segad et al. (2010) make the above quoted statement in the beginning of the paper, they only conclude that “tactoids” are formed in pure Ca-montmorillonite.
The swelling and sedimentation behavior of Ca-montmorillonite is a very interesting question, that we do not have all the answers to yet. Still, it is basically irrelevant for making statements about the structure in compacted — sodium dominated3 — bentonite.
Churakov et al. (2014) also give two references for the statement that the “interparticle porosity” in montmorillonite is 10 – 30% of the total porosity: Holmboe et al. (2012) and Kozaki et al. (2001). This is a bizarre way of referencing, as these two studies draw incompatible conclusions, and since Holmboe et al. (2012) — which is the more adequately performed study — state that this type of porosity may be absent:
At dry density \(>1.4 \;\mathrm{g/cm^3}\) , the average interparticle
porosity for the [natural Na-dominated bentonite and purified
Na-montmorillonite] samples used in this study was found to be
\(1.5\pm1.5\%\), i.e. \(\le 3\%\) and significantly lower than
previously reported in the literature.
Holmboe et al. (2012)
address directly the discrepancy with earlier studies, and suggest
that these were not properly analyzed
The apparent discrepancy between the basal spacings reported by Kozaki et al. (1998, 2001) using Kunipia-F washed Na-montmorillonite, and by Muurinen et al. (2004), using a Na-montmorillonite originating from Wyoming Bentonite MX-80, and the corresponding average basal spacings of the [Na-montmorillonite originating from Wyoming bentonite MX-80] samples reported in this study may partly be due to water contents and partly to the fact that only apparent \(\mathrm{d_{001}}\) values using Bragg’s law, without any profile fitting, were reported in their studies.
If
Kozaki et al. (2001)
should be used to support a claim about “interparticle porosity”, it
consequently has to be done in opposition to — not in conjunction
with —
Holmboe et al. (2012).
It would then also be appropriate for authors to provide arguments for
why they discard the conclusions of
Holmboe et al. (2012).4
Stacks in compacted bentonite make no geometrical sense
The literature is full of fancy figures of bentonite structure involving stacks. A typical example is found in Wu et al. (2018), and looks similar to this:
This illustration is part of a figure with the caption “Schematic representation of the different porosities in bentonite and the potential diffusion paths.”5 The regular rectangles in this picture illustrate stacks that each seems to contain five TOT-layers (I assume this throughout). Conveniently, these groups of five layers have the same length within each stack, while the length varies somewhat between stacks. This is a quite common feature in figures like this, but it is also common that all stacks are given the same length.
Another feature this illustration has in common with others is that the particles are ordered: we are always shown edges of the TOT-layers. I guess this is partly because a picture of a bunch of stacks seen from “the top” would be less interesting, but it also emphasizes the problem of representing the third dimension: figures like these are in practice figures of straight lines oriented in 2D, and the viewer is implicitly required to imagine a 3D-version of this two-dimensional representation.
A “realistic” stack picture
But, even as a 2D-representation, these figures are not representative
of what an actual configuration of stacks of TOT-layers looks like.
Individual TOT-layers have a distinct thickness of about 1 nm, but
varies widely in the other two dimensions.
Ploehn and Liu (2006)
analyzed the size distribution of Na-montmorillonite (“Cloisite
Na+”) using atomic force microscopy, and found an average aspect
ratio of 180 (square-root of basal area divided by thickness). A
representative single “TOT-line” drawn to scale is consequently
quite different from what is illustrated in in most stack-pictures,
and look like this (click on the figure to see it in full size)
In this figure, we have added “water layers” on each side of the TOT-layer (light red), with the water-to-solid volume ratio of 16. Neatly stacking five such units shows that the rectangles in the Wu et al. (2018)-figure should be transformed like this
But this is still not representative of what an assemblage of five
randomly picked TOT-layers would look like, because the size
distribution has a substantial variance. According to
Ploehn and Liu (2006), the
aspect ratio follows approximately a log-normal distribution. If we
draw five values from this distribution for the length of five
“TOT-lines”, and form assemblages, we end up with structures that
look like this:7
These are the kind of units that should fill the bentonite illustrations. They are quite irregularly shaped and are certainly not identical (this would be even more pronounced when considering the third dimension, and if the stacks contain more layers).
It is easy to see that it is impossible to construct a dense structure
with these building blocks, if they are allowed a random
orientation. The resulting structure rather looks something like this
Such a structure evidently has very low density, and are reminiscent of the gel structures suggested in e.g. Shalkevich et al. (2007) (see fig. 7 in that paper). This makes some sense, since the idea of stacks of TOT-layers (“tactoids”) originated from studies of low density structures, as discussed above.
Note that the structure in pictures like that in Wu et al. (2018) has a substantial density only because it is constructed with stacks with an unrealistic shape. But even in these types of pictures is the density not very high: with some rudimentary image analysis we conclude that the density in the above picture is only around 800 kg/m3. Also the figure from Navarro et al. (2017) above gives a density below 1000 kg/m3, although there it is explicitly stated that it is a representation of “highly compacted bentonite”.
The only manner in which the “realistic” building blocks can be
made to form a dense structure is to keep them in the same
orientation. The resulting structures then look e.g. like this
where we have color coded each stack, to remind ourselves that these
units are supposed to be fundamental.
Just looking at this structure of a “stack of stacks” should make it clear how flawed the idea is of stacks as fundamental structural units in compacted bentonite (note also how unrepresentative the stack-pictures found in the literature are). One of many questions that immediately arises is e.g. why on earth the tiny gaps between stacks (indicated by arrows) should remain. This brings us to the next argument against stacks as fundamental units for compacted water saturated bentonite:
What is supposed to keep stacks together?
Compacted bentonite of interest e.g. for sealing in radioactive waste repositories exerts swelling pressure of several MPa when in contact with external water. This osmotic pressure is a consequence of the presence of the mobile exchangeable cations in montmorillonite. Each “realistic” unit that we have imagined above is thus required to be at a huge elevated pressure, and the individual TOT-layers have a strong driving force to separate. And, unless a mechanism is provided for why such a separation is impossible, this is of course what we expect to happen! As far as I am aware, such a separation inhibiting mechanism has never been suggested in any publication that promotes the stack concept in compacted bentonite. To get a feel for the absurdity of this issue, let’s take a new look at the figure from Navarro et al. (2017)
Assuming that this system is in equilibrium with an external water
reservoir at zero pressure (i.e. atmospheric absolute pressure), the
pressure in the compartment labeled “intra-aggregate space” is also
close to zero. On the other hand, in the “stacks” located just a few
nm away, the pressure is certainly above 10 MPa in many places! A
structure like this is obviously not in mechanical equilibrium! (To use
the term “obvious” here feels like such an understatement.)
Implications
To sum up what we have discussed so far, the following picture
emerges. The bentonite literature is packed with descriptions of
compacted water saturated bentonite as built up of stacks as
fundamental units. These descriptions are so commonplace that they
often are not supported by references. But when they are, it seems
that the entire notion is based on misconceptions. In particular,
structures identified in low density systems (suspensions, gels) have
been carried over, without reflection, to descriptions of compacted
bentonite. Moreover, all figures illustrating the stack concept are
based on inadequate representations of what an arbitrary assemblage of
TOT-layers arranged in this way actually would look like. With a
“realistic” representation it quickly becomes obvious that it makes
little sense to base a fundamental unit in compacted systems on the
stack concept.
My impression is that this flawed stack concept underlies the entire
contemporary mainstream view of compacted bentonite, as e.g. expressed
by
Wu et al. (2018):
A widely accepted view is that the total porosity of bentonite
consists of \(\epsilon_ {ip}\) and \(\epsilon_ {il}\) (Tachi and
Yotsuji, 2014; Tournassat and Appelo, 2011; Van Loon et al., 2007).
\(\epsilon_ {ip}\) is a porosity related to the space between the
bentonite particles and/or between the other grains of minerals
present in bentonite. It can further be subdivided into
\(\epsilon_ {ddl}\) and \(\epsilon_ {free}\). The diffuse double layer,
which forms in the transition zone from the mineral surface to the
free water space, contains water, cations and a minor amount of
anions. The charge at the negative outer surface of the
montmorillonite is neutralized by an excess of cations. The free
water space contains a charge-balanced aqueous solution of cations
and anions. \(\epsilon_ {il}\) represents the space between TOT-layers
in montmorillonite particles exhibiting negatively charged
surfaces. Due to anion exclusion effect, anions are excluded from
the interlayer space, but water and cations are present.
This view can be summarized as:
The fundamental building blocks are stacks of TOT-layers
(“particles”, “aggregates”, “tactoids”, “grains”…)
Electric double layers are present only on external
surfaces of the stacks.
Far away from external surfaces — in the “inter-particle” or
“inter-aggregate” pores — the diffuse layers merge with a bulk
water solution
Interlayer pores are defined as being internal to the stacks,
and are postulated to be fundamentally different from the external
diffuse layers; they play by a different set of rules.
I don’t understand how authors can get away with promoting this
conceptual view without supplying reasonable arguments for all of its
assumptions8 — and with such a
complex structure, there are a lot of assumptions.
As already discussed, the geometrical implications of the suggested structure do not hold up to scrutiny. Likewise, there are many argumentsagainst the presence of substantial amounts of bulk water in compacted bentonite, including the pressure consideration above. But let’s also take a look at what is stated about “interlayers” and how these are distinguished from electric double layers (I will use quotation marks in the following, and write “interlayers” when specifically referring to pores defined as internal to stacks).
“Interlayers”
“Interlayers” are often postulated to be completely devoid of anions. We discussed this assumption in more depth in a previous blog post, where we discovered that the only references supplied when making this postulate are based on the Poisson-Boltzmann equation. But this is inadequate, since the Poisson-Boltzmann equation does describe diffuse layers, and predicts anions everywhere.
By requiring anion-free “interlayers”, authors actually claim that the physico-chemistry of “interlayers” is somehow qualitatively different from that of “external surfaces”, although these compartments have the exact same constitution (charged TOT-layer surface + ions + water). But an explanation for why this should be the case is never provided, nor is any argument given for why diffuse layer concepts are not supposed to apply to “interlayers”.9 This issue becomes even more absurd given the strong empirical evidence for that anions actually do reside in interlayers.
The treatment of anions is not the only ad hoc description of “interlayers”. It also seems close to mandatory to describe them as having a maximum extension, and as having an extension independently parameterized by sample density. E.g. the influential models for Na-bentonite of Bourg et al. (2006) and Tournassat and Appelo (2011) both rely on the idea that “interlayers” swell out to a certain volume that is smaller than the total pore volume, but that still depends on density.
In e.g. Bourg et al. (2006), the fraction of “interlayer” pores remains essentially constant at ~78%, as density decreases from 1.57 g/cm3 to 1.27 g/cm3, while the “interlayers” transform from having 2 monolayers of water (2WL) to having 3 monolayers (3WL). This is a very strange behavior: “interlayers” are acknowledged as having a swelling potential (2WL expands to 3WL), but do, for some reason, not affect 22% of the pore volume! Although such a behavior strongly deviates from what we expect if “interlayers” are treated with conventional diffuse layer concepts, no mechanism is provided.
Another type of macabre consequence of defining “interlayer” pores as internal to stacks is that a completely homogeneous system is described has having no interlayer pores (because it has no stacks). E.g. Tournassat and Appelo (2011) write (\(n_c\) is the number of TOT-layers in a stack)
[…] the number of stacks in the \(c\)-direction has considerable influence on the interlayer porosity, with interlayer porosity increasing with \(n_c\) and reaching the maximum when \(n_c \approx 25\). The interlayer porosity halves with \(n_c\) when \(n_c\) is smaller than 3, and becomes zero for \(n_c = 1\).10
It is not acceptable that using the term interlayer should require
accepting stacks as fundamental units. But the usage of the term as
being internal to stacks is so widespread in the contemporary
bentonite literature, that I fear it is difficult to even communicate
this issue. Nevertheless, I am certain that e.g.
Norrish (1954) does not
depend on the existence of stacks when using the term like this:
Fig. 7 shows the relationship between interlayer spacing and water
content for Na-montmorillonite. There is good agreement between the
experimental points and the theoretical line, showing that
interlayer swelling accounts for all, or almost all, of physical
swelling.
The stack view obstructs real discovery
A severe consequence of the conceptual view just discussed is that “stacking number” — the (average) number of TOT-layers that stacks are supposed to contain — has been established as fitting parameter in models that are clearly over-parameterized. An example of this is Tournassat and Appelo (2011), who write11
Our predictive model excludes anions from the interlayer space and
relates the interlayer porosity to the ionic strength and the
montmorillonite bulk dry density. This presentation offers a good
fit for measured anion accessible porosities in bentonites over a
wide range of conditions and is also in agreement with microscopic
observations.
But since anions do reside in interlayers,12 it would be better if the model didn’t fit: an over-parameterized or conceptually flawed model that fits data provides very little useful information.
A similar more recent example is Wu et al. (2018). In this work, a model based on the stack concept is successfully fitted both to data on \(\mathrm{ReO_4^-}\) diffusion in “GMZ” bentonite and to data on \(\mathrm{Cl^-}\) diffusion in “KWK” bentonite, by varying “stacking number” (among other parameters). Again, as the model assumes anion-free “interlayer” pores, a better outcome would be if it was not able to fit the data. Moreover, this paper focuses mainly on the ability of the model, while not at all emphasizing the fact that about ten (!) times more \(\mathrm{ReO_4^-}\) was measured in “GMZ” as compared with \(\mathrm{Cl^-}\) in “KWK”, at similar conditions in certain cases. The latter observation is quite puzzling and is, in my opinion, certainly worth deeper investigation (and I am fully convinced that it is not explained by differences in “stacking number”).
[3] Note that “sodium
dominated” in this context means ~20% or more.
[4] It may be noticed that Kozaki et al. (2001) see no X-ray diffraction peaks for low density samples:
The basal spacing of water-saturated
montmorillonite was determined by the XRD method. […] It was found
that a basal spacing of 1.88 nm, corresponding to the three-water
layer hydrate state […] was not observed before the dry density
reached 1.0 Mg/m3.
My interpretation of this observation is that the diffraction peak has
shifted to even lower reflection angles (in agreement with the
observations
of Holmboe
et al. (2012)), not registered by the equipment. The alternative
interpretation must otherwise be that “stacks” suddenly cease to
exist below 1.0 g/cm3. (Yet,
Kozaki et al. (2001)
continues to use a certain d-value in their analysis, also for densities
below 1.0 g/cm3.)
[5] I have discussed “diffusion
paths” in an
earlier blog post.
This illustration certainly fits that discussion.
[6] A water-to-solid volume ratio of 1 corresponds basically to
interlayers of three monolayers of water (3WL).
[7] To construct these units, I made the additional choice of placing each layer randomly in the horizontal direction, with the constraint that all layers should be confined within the range of the longest one in each unit.
[8] By “get away with” I mean “pass peer-review”, and by “don’t understand” I mean “understand”.
[10] A mathematical remark: if the interlayer porosity “halves with \(n_c\)” (what does that mean?) when \(n_c = 2\) (“smaller than 3”), it is impossible to simultaneously have zero interlayer porosity for \(n_c = 1\) (unless the interlayer porosity is zero for any \(n_c\)).
[11] I guess the word “presentation” here really should be “representation”?
[12] Note that one of the authors of this paper also claims in a later paper that anions do populate 3-waterlayer interlayers, in accordance with the Poisson-Boltzmann equation:
The agreement
between PB calculations and MD simulation predictions was somewhat
worse in the case of the \(\mathrm{Cl^-}\) concentration profiles than
in the case of the \(\mathrm{Na^+}\) profiles (Figure 3), perhaps
reflecting the poorer statistics for interlayer Cl concentrations
[…] Nevertheless, reasonable quantitative agreement was found
(Table 2).

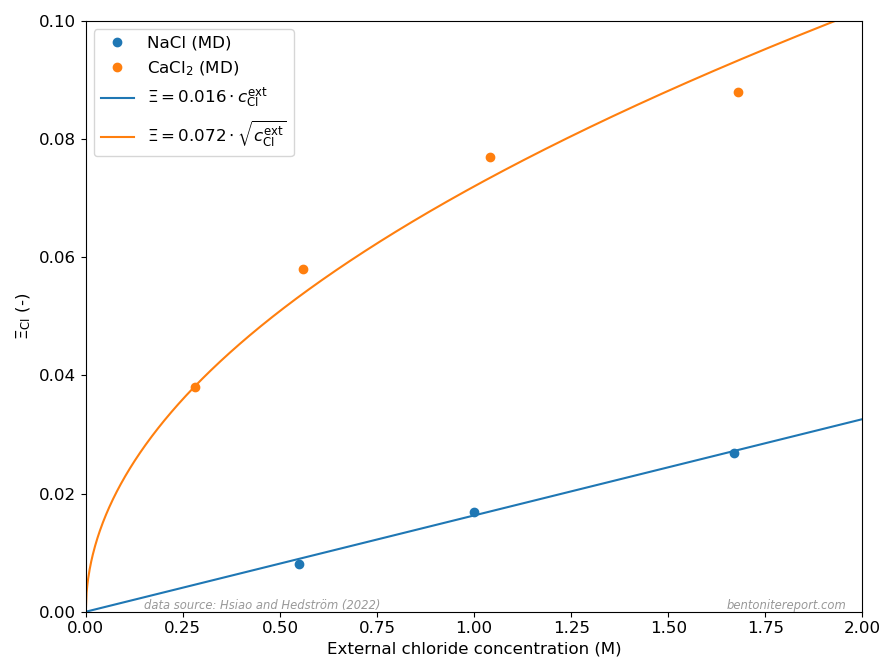
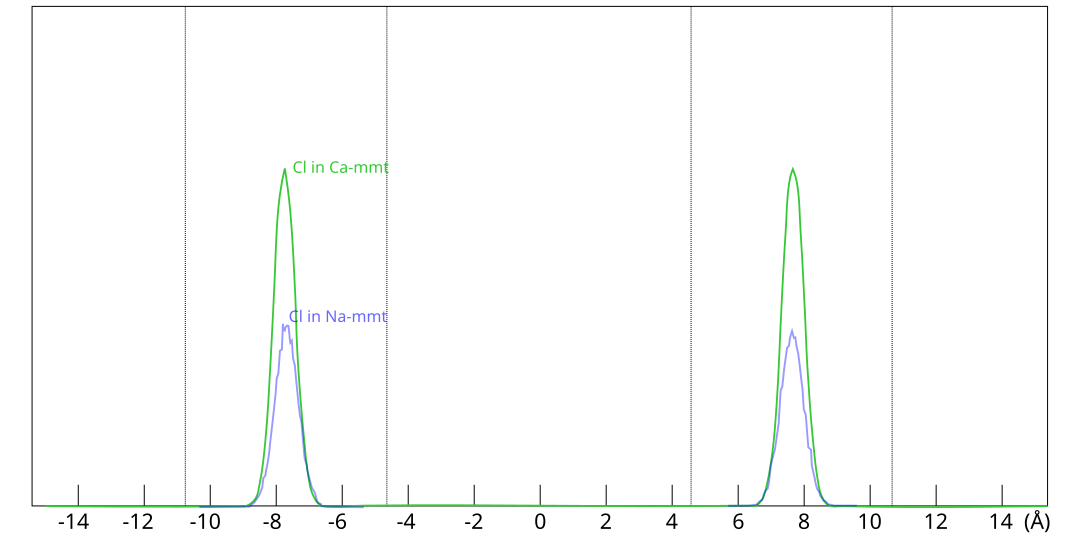












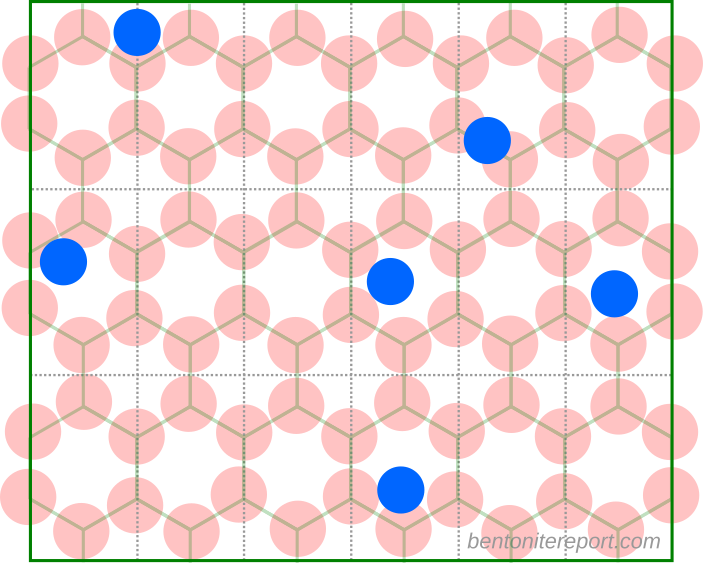
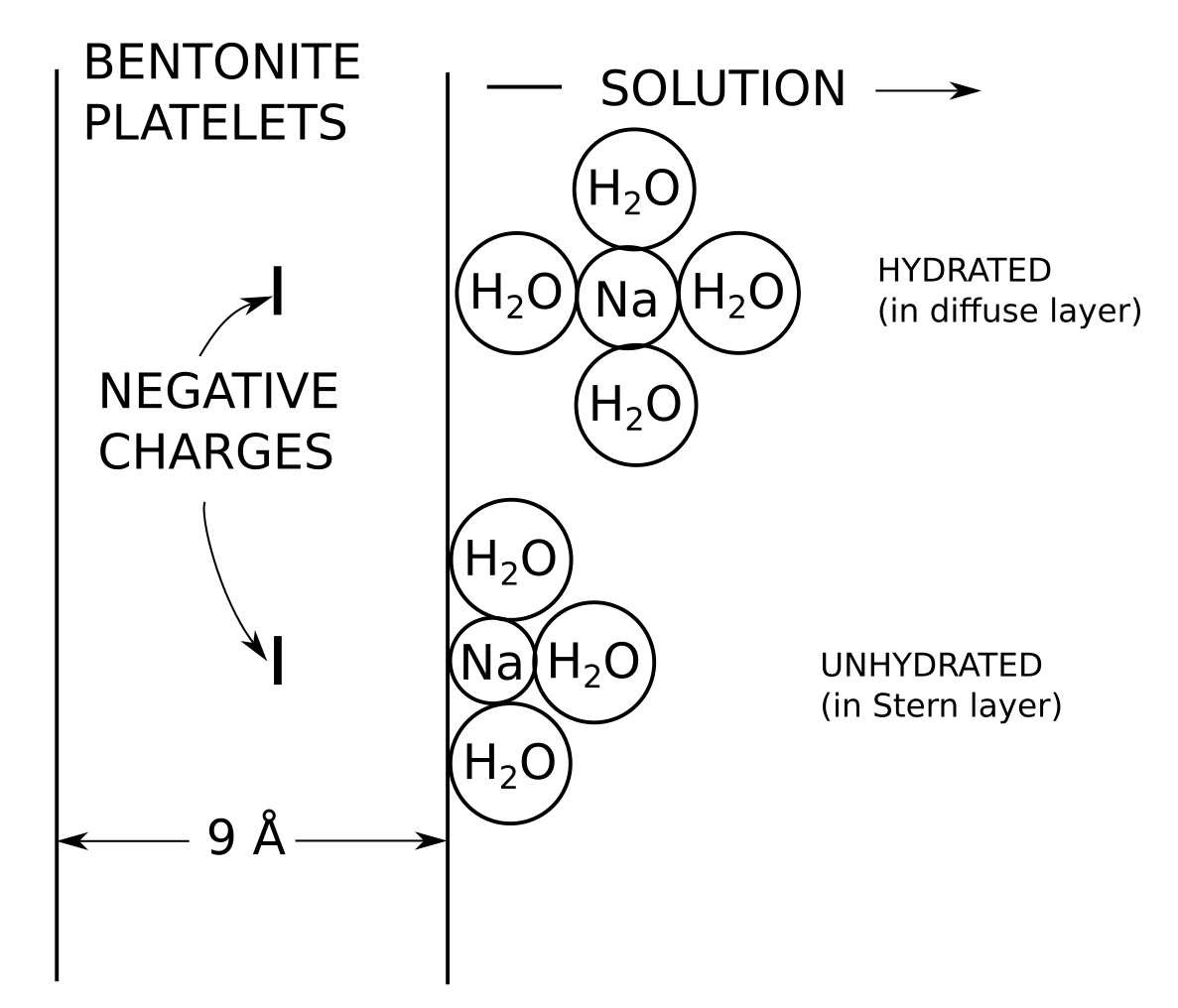

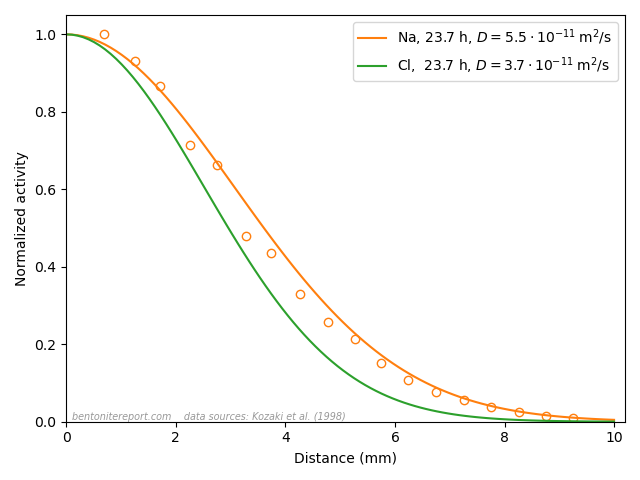




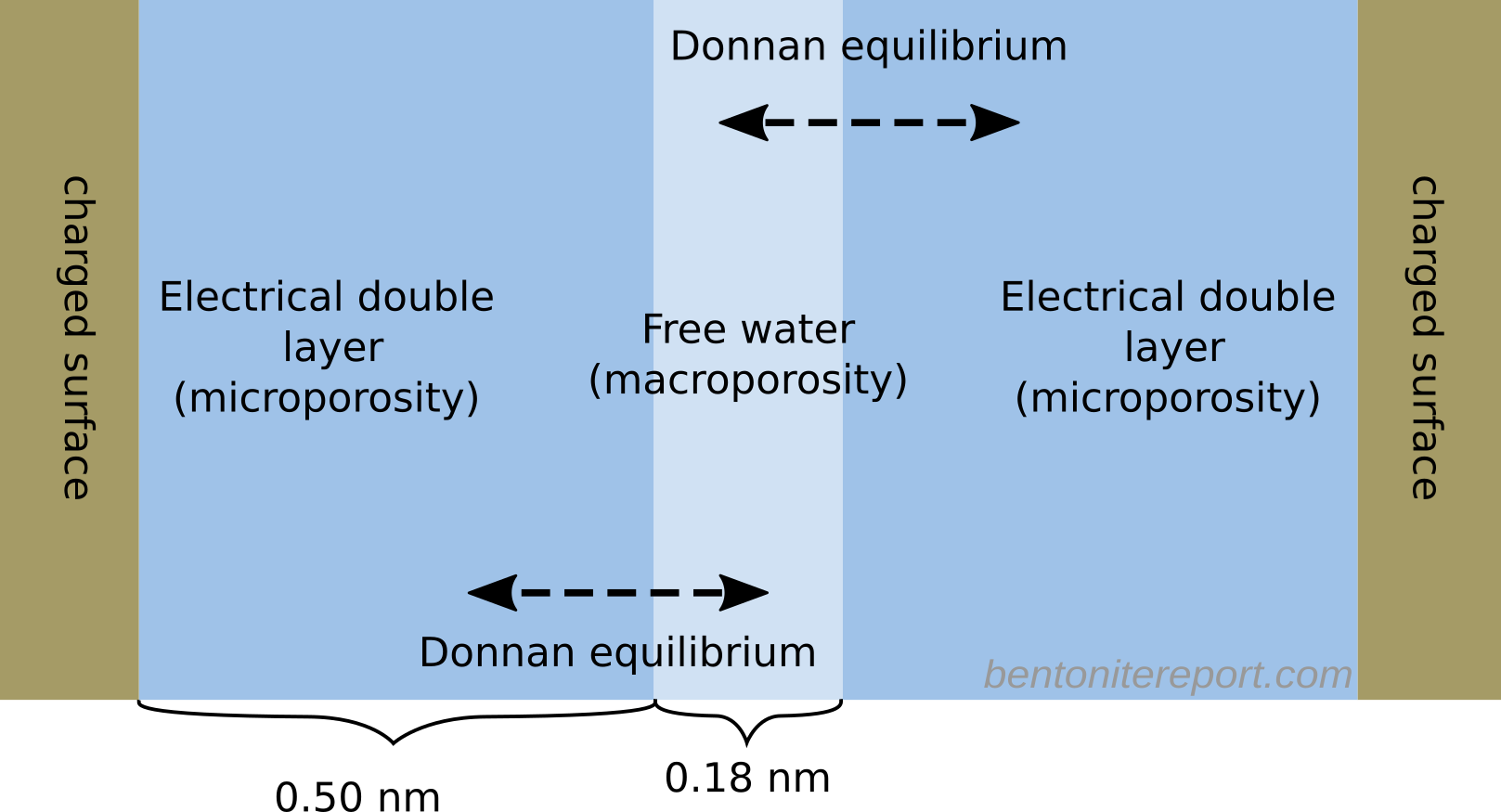






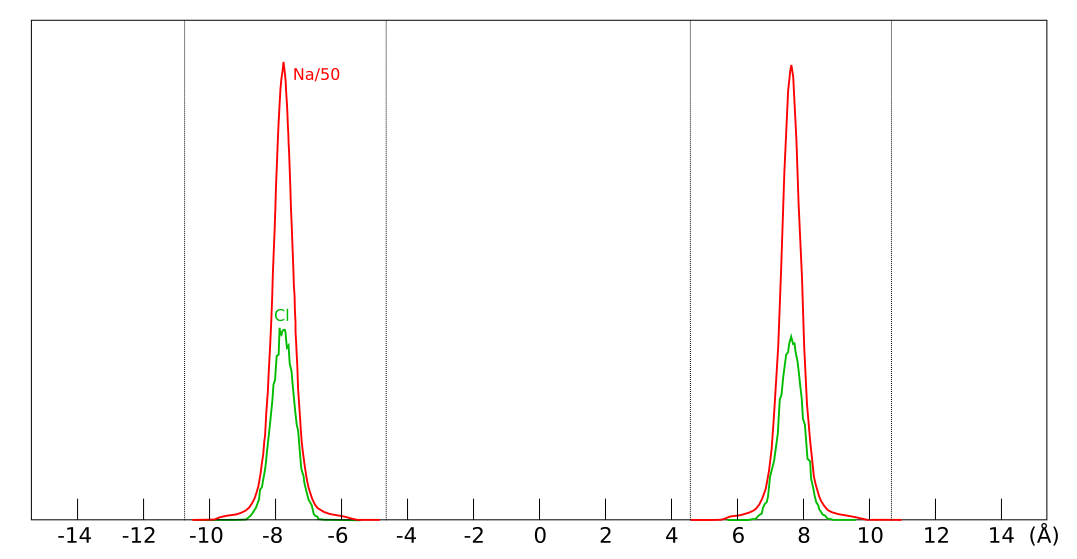












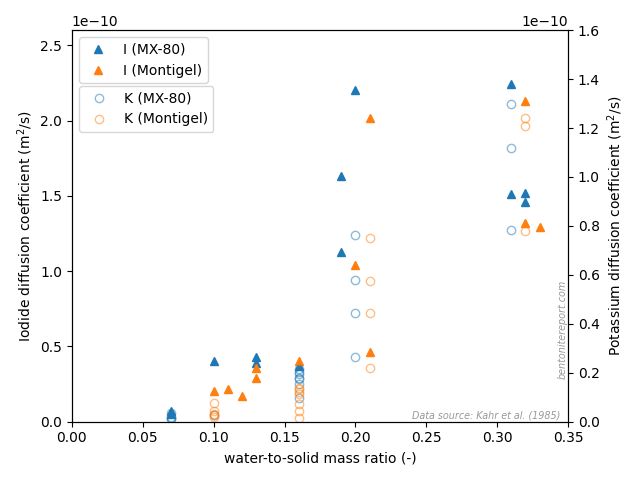

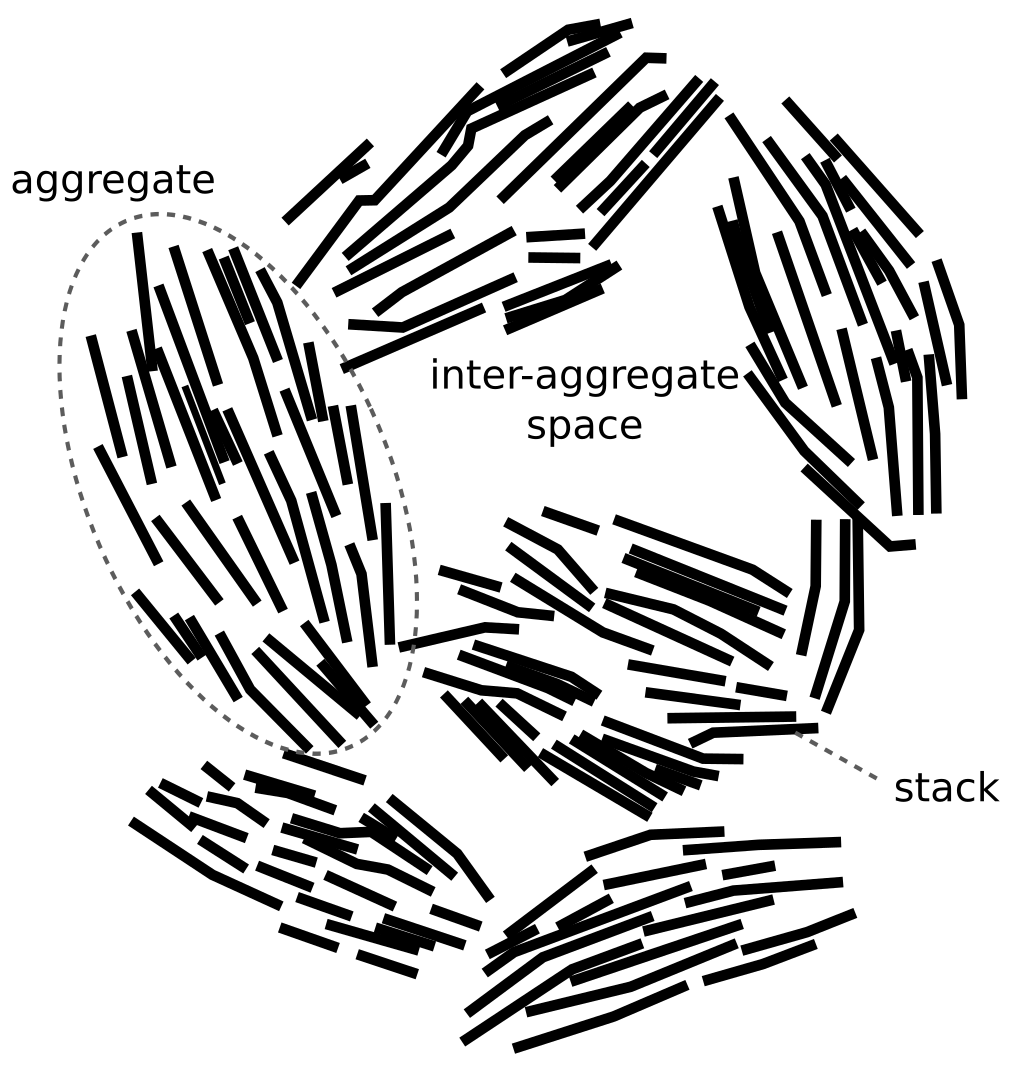







{kind=link}
{kind=link}