Repulsion between surfaces and anions is not really the
point
Many publications dealing with “anion” exclusion in compacted bentonite describe the phenomenon as being primarily due to electrostaticrepulsion of anions from the negativelychargedclaysurfaces. This explanation, which may seem plausible both at a first and a second glance, is actually not that satisfactory. There are two major issues to consider:
Although it is popular to use the word “anion” when referring to the phenomenon, it must be remembered that the anions are accompanied by cations, in order to maintain overall charge neutrality; it really is salt that is excluded from the bentonite. This observation shows that the above “explanation” is incomplete: it can be argued with the same logic that salt should accumulate, because the clay surfaces attract the cations of the external salt.
Salt exclusion occurs generally in Donnan systems, also in those that lack surfaces. Its principal explanation can consequently not involve the presence of surfaces. For a simpler system, e.g. potassium ferrocyanide, the “explanation” above translates to claiming that exclusion is caused by “anions” being electrostatically repelled by the ferrocyanide ions. In this case it may be easier to spot the shortcoming of such a claim, and to consider also the potassium ions (which attract anions), as well as the role played by the cations of the excluded salt.
What, then, is the primary cause for salt exclusion? Let us continue with using potassium ferrocyanide as an example of a simple Donnan system, and then translate our findings to the case of compacted bentonite.
Ferrocyanide
Consider a potassium ferrocyanide solution separated from a potassium
chloride solution by a membrane permeable to all but the ferrocyanide
ions. The ionic configuration near the membrane then looks something
like this
Because potassium ions can pass the membrane, and because they have an entropic driving force to migrate out of the ferrocyanide solution, a (microscopic) region is formed in the external solution next to the membrane, with an excess amount of positive charge. Similarly, a region is formed next to the membrane in the ferrocyanide solution with an excess amount of negative charge. Thus, a region of charge separation exists across the membrane — similar to the depletion zone in a p-n junction — over which the electrostatic potential varies. The electric field (= a varying potential) at the interface acts as to pull back potassium ions towards the ferrocyanide solution. The equilibrium width of the space charge region is set when the diffusive flux is balanced by the flux due to the electric field.
With a qualitative understanding of the electrostatic potential configuration we can now give the most plain answer to what causes “anion” exclusion: it is because of the potential difference across the membrane. Chloride ions behave in the opposite way as compared to potassium, with an entropic driving force to enter the ferrocyanide solution, while being pulled back towards the external solution due to the electric field across the membrane.
Here the mindful reader may perhaps object and point out that the electric field restricting the chloride inflow reasonably originates from the ferrocyanide anions. It thus may seem that “anion” exclusion, after all, is caused by repulsion from other negative charges.
Indeed, electrostatic repulsion of anions requires the “push” of some other negatively charged entity. But note that the potential is constant in the interior of the ferrocyanide solution, and only varies near the membrane. The variation of the potential is caused by separation of charge: chloride is as much “pushed” out of the ferrocyanide solution by the ferrocyanide as it is “pulled” out of it, due to electrostatic attraction, by the excess potassium on the other side. Repulsion between charges of equal sign occurs also in the interior of the ferrocyanide solution (or in any ionic solution), but does not in itself lead to salt exclusion.
Bentonite
The above description can be directly transferred to the case of compacted bentonite. Replacing the potassium ferrocyanide with e.g. K-montmorillonite, salt exclusion occurs mainly because potassium can migrate out of the clay region, while montmorillonite particles cannot. Again, we have charge separation with a resulting varying electrostatic potential across the interface.
Admittedly, the general situation is more complicated in bentonite because of the extension of montmorillonite particles; viewed as “anions”, these are irregularly shaped macromolecules with hundreds or thousands of charge centers.
The ion configuration in a bentonite suspension therefore looks quite different from a corresponding ordinary solution, as the montmorillonite charge obviously is constrained to individual particles. Dilute systems thus have charge separation on the particle scale and show salt exclusion even without charge separation at the interface to the external solution. These types of systems (suspensions) have historically been the subject of moststudies on “anion”exclusion, and are usually treated theoretically using the Gouy-Chapman model.
With increasing density, however, the effect of a varying potential between montmorillonite particles diminishes, while the effect of charge separation at the interface increases. For dense systems (> 1.2 g/cm3, say), we may therefore approximate the internal potential as constant and only consider the variation across the interface to the external solution using Donnan’s “classical” framework.1
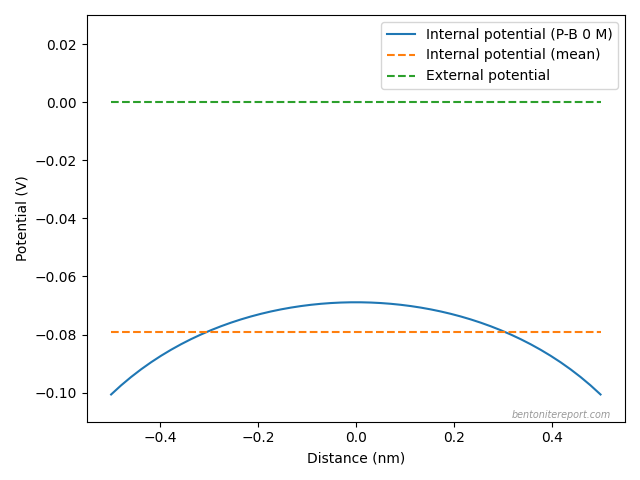
Here is an illustration of the validity of this approximation:
The figure shows the difference between the external (green) and the average internal (orange) potentials in a 1:1 system of density 1.3 g/cm3 and with external concentration 0.1 M, calculated using Donnan’s “classical” equation. Also plotted is the electrostatic potential across the interlayer (blue) as calculated using the Poisson-Boltzmann equation,2 in a similar system (interlayer distance 1 nm). It is clear that the variation of the Poisson-Boltzmann potential from the average is small in comparison with the Donnan potential.
Repulsion between chloride and montmorillonite particles of course occurs everywhere in compacted bentonite, whereas the phenomenon mainly responsible for salt exclusion occurs only near the interfaces. Merely stating electrostatic repulsion as the cause for salt exclusion in compacted bentonite does not suffice, just as in the case of ferrocyanide.
To illustrate that the salt exclusion effect depends critically on exchangeable cations being able to diffuse out of the bentonite, consider the following thought experiment.3 Compacted K-montmorillonite is contacted with a NaCl solution. But rather than having a conventional component separating the solution and the clay, we imagine a membrane that does not allow for the passage of neither potassium nor clay, but that allows for the passage of sodium and chloride. Since potassium is not allowed to diffuse out of the bentonite, no charge separation occurs across the membrane. With no space charge region, the electrostatic potential does not vary and NaCl is not excluded! (to the extent that the Donnan approximation is valid)
A charge neutral perspective
The explanation for “anion” exclusion that we have explored rests on
the formation of a potential difference across the interface region
between bentonite and external solution. But remember that it is salt
— in our example KCl — that is excluded from the bentonite (or the
ferrocyanide solution), and that the cation (K) gains energy by being
transferred from the external to the internal solution. The electrical
work for transferring a unit of KCl is thus zero (which makes sense
since KCl is a charge neutral entity). In this light, it may seem
unsatisfactory to offer the potential difference as the sole
explanation for salt exclusion.
I therefore think that the following kinematic way of reasoning is very helpful. Instead of considering the mass transfer of Cl across the membrane in terms of oppositely directed “electric” and “diffusive” parts, we lump them together with equal amounts of K transfer, giving two equal but oppositely directed fluxes of KCl. Reasonably, the KCl flux into the ferrocyanide solution is proportional to the external ion concentrations
\(A\) is a coefficient accounting for the transfer resistance across the interface region. Requiring the sum of these fluxes to be zero gives the following relation
We can therefore interpret KCl exclusion as an effect of potassium in the clay providing a potential for “out-transfer”, as soon as the chance is given, i.e. when chloride enters from the external solution. From this perspective salt exclusion could maybe be said to be a form of cation “rejection”.
Footnotes
[1]
Note also that the Gouy-Chapman model is not valid in the
high density limit, although it is
applied (or
alluded to)
in this limit in
manypublications.
But e.g. Schofield (1947)
states (about the Gouy-Chapman solution):
[T]he equation is applicable to cases in which the distance
between opposing surfaces considerably exceeds the distance
between neighboring point charges on the surfaces; for there
will then be a range of electrolyte concentrations over which
the radius of the ionic atmosphere is less than the former and
greater than the latter.
This criterion is not met in compacted bentonite, where instead the interlayer distance is comparable to the distance between neighboring charge centers on the surfaces. Invalid application of the Gouy-Chapman model also seems to underlie the flawed but widespread “anion-accessible porosity” concept.
[2] This calculation uses the equations presented in Engström and Wennerström (1978), and assumes no excess ions and a surface charge density of 0.111 \(\mathrm{C/m^2}\). For real consistency this calculation should really be performed with the boundary condition of 0.1 M external concentration. However, since the purpose of the graph is just to demonstrate the sizes of the two potential variations, and since I have yet to acquire a reasonable tool for performing Poisson-Boltzmann calculations with non-zero external concentration, I disregard this inconsistency. Moreover, the continuum assumption of the Poisson-Boltzmann description is anyway beginning to lose its validity at these interlayer distances. Update (220831): Solutions to the Poisson-Boltzmann equation with non-zero external concentration are presented here.
[3] Perhaps this could be done as a Molecular Dynamics
simulation?
At the atomic level, montmorillonite is built up of so-called TOT-layers: covalently bonded sheets of aluminum (“O”) and silica (“T”) oxide (including the right amount of impurities/defects). In my mind, such TOT-layers make up the fundamental particles of a bentonite sample. Reasonably, since montmorillonite TOT-layers vary extensively in size, and since a single cubic centimeter of bentonite contains about ten million billions (\(10^{16}\)), they are generally configured in some crazily complicated manner.
Stack descriptions in the literature
But the idea that the single TOT-layer is the fundamental building
block of bentonite is not shared with many of today’s bentonite
researchers. Instead, you find descriptions like e.g. this one, from
Bacle et al. (2016)
Clay mineral particles consist of stacks of parallel
negatively-charged layers separated by interlayer
nanopores. Consequently, compacted smectite contains two major
classes of pores: interlayer nanopores (located inside the
particles) and larger mesopores (located between the particles).
In compacted rocks, montmorillonite (Mt) forms aggregates
(particles) with 5–20 TOT layers (Segad et al., 2010). A typical
radial size of these particles is of the order of 0.01 to 1
\(\mathrm{\mu m}\). The pore space between Mt particles is referred to
as interparticle porosity. Depending on the degree of compaction,
the interparticle porosity contributes 10 to 30% of the total water
accessible pore space in Mt (Holmboe et al., 2012; Kozaki et al.,
2001).
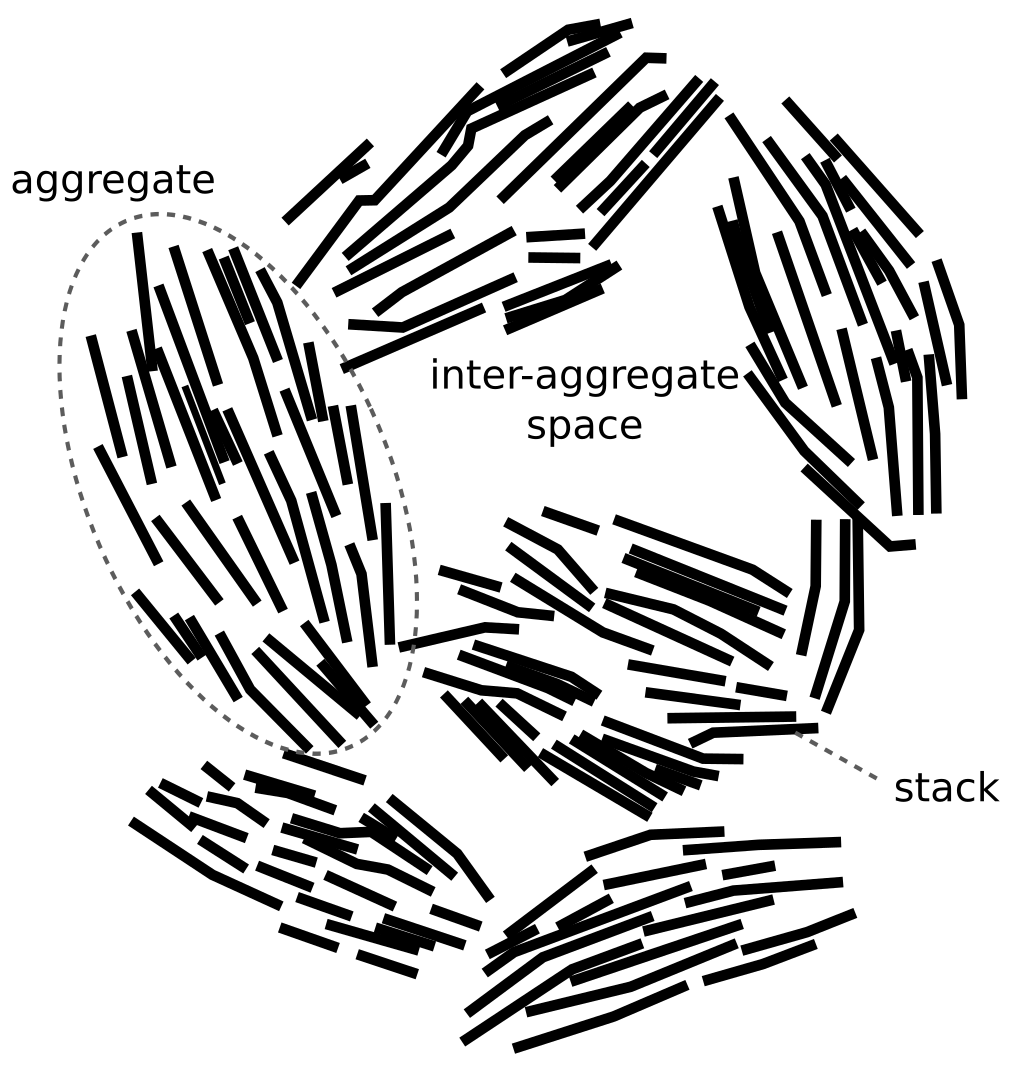
Such statements show that researchers have something more complex in mind than individual TOT-layers when speaking of “particles”: they are some sort of assemblages of TOT-layers. The quotation of Bacle et al. (2016), using both the terms “stacks” and “particles”, even hints at an idea of a hierarchy of fundamental structures. Such a hierarchy is expressed explicitly in e.g. Navarro et al. (2017), who provide a figure with the caption “Schematic particle arrangement in highly compacted Na-bentonite” that looks similar to this one:
Here it is clear that they differ between “aggregates” (which I’m
not sure is the same thing as “particles”), “stacks”, and
individual TOT-layers (which I assume are represented by the
line-shaped objects). In the following, however, we will use the term
“stack” to refer to any kind of suggested fundamental structure
built up from individual TOT-layers.
The one-sentence version of this blog post is:
Stacks make no sense as fundamental building blocks in models of water saturated, compacted bentonite.
The easiest argument against stacks is, in my mind, to simply work out
the geometrical consequences. But before doing that we will examine
some of the references given to support statements about stacks in
compacted systems. Often, no references are given at all, but when
they are, they usually turn out to be largely irrelevant for the
system under study, or even to support an opposite view.
Inadequate referencing
As an example (of many) of inadequate referencing, we
use the statement above from Churakov et al. (2014) as
a starting point. I think this is a “good” statement, in the sense
that it makes rather precise claims about how compacted bentonite is
supposed to be structured, and provides references for some key
statements, which makes it easier to criticize.
Clay is normally not a homogeneous lamellar material. It might be
better described as a disordered structure of stacks of platelets,
sometimes called tactoids — a tactoid typically consists of 5-20
platelets.19-21
Here the terminology is quite different from the previous quotations: TOT-layers are called “platelets”, and “particles” are called “tactoids”. Still, they use the phrase “stacks of platelets”, so I think we can continue with using “stack” as a sort of common term for what is being discussed.1 We may also note that here is used the word “clay”, rather than “montmorillonite” (as does Bacle et al. (2016)), but it is clear from the context of the article that it really is montmorillonite/bentonite that is discussed.
Anyhow, Segad et al. (2010) do not give much direct information on the claim we investigate, but provide three new references. Two2 of these — Banin (1967) and Shalkevich et al. (2007) — are actually studies on montmorillonite suspensions, i.e. as far away as you can get from compacted bentonite in terms of density; the solid mass fraction in these studies is in the range 1 – 4%.
The average distance between individual TOT-layers in this density limit is comparable with, or even larger than, their typical lateral extension (~100 nm). Therefore, much of the behavior of low density montmorillonite depends critically on details of the interaction between layer edges and various other components, and systems in this density limit behave very differently depending on e.g. ionic strength, cation population, preparation protocol, temperature, time, etc. This complex behavior is also connected with the fact that pure Ca-montmorillonite does not form a sol, while the presence of as little as 10 – 20% sodium makes the system sol forming. The behaviors and structures of montmorillonite suspensions, however, say very little about how the TOT-layers are organized in compacted bentonite.
We have thus propagated from a statement in Churakov et al. (2014), and a similar one in Segad et al. (2010), that montmorillonite in general, in “compacted rocks” forms aggregates of 5 – 20 TOT-layers, to studies which essentially concern different types of materials. Moreover, the actual value of “5 – 20 TOT layers” comes from Banin (1967), who writes
Evidence has accumulated showing that when montmorillonite is
adsorbed with Ca, stable tactoids, containing 5 to 20 parallel
plates, are formed (1). When the mineral is adsorbed with Na, the
tactoids are not stable, and the single plates are separated from
each other.
This source consequently claims that the single TOT-layers are the fundamental units, i.e. it provides an argument against any stack concept! (It basically states that pure Ca-montmorillonite does not form a sol.) In the same manner, even though Segad et al. (2010) make the above quoted statement in the beginning of the paper, they only conclude that “tactoids” are formed in pure Ca-montmorillonite.
The swelling and sedimentation behavior of Ca-montmorillonite is a very interesting question, that we do not have all the answers to yet. Still, it is basically irrelevant for making statements about the structure in compacted — sodium dominated3 — bentonite.
Churakov et al. (2014) also give two references for the statement that the “interparticle porosity” in montmorillonite is 10 – 30% of the total porosity: Holmboe et al. (2012) and Kozaki et al. (2001). This is a bizarre way of referencing, as these two studies draw incompatible conclusions, and since Holmboe et al. (2012) — which is the more adequately performed study — state that this type of porosity may be absent:
At dry density \(>1.4 \;\mathrm{g/cm^3}\) , the average interparticle
porosity for the [natural Na-dominated bentonite and purified
Na-montmorillonite] samples used in this study was found to be
\(1.5\pm1.5\%\), i.e. \(\le 3\%\) and significantly lower than
previously reported in the literature.
Holmboe et al. (2012)
address directly the discrepancy with earlier studies, and suggest
that these were not properly analyzed
The apparent discrepancy between the basal spacings reported by Kozaki et al. (1998, 2001) using Kunipia-F washed Na-montmorillonite, and by Muurinen et al. (2004), using a Na-montmorillonite originating from Wyoming Bentonite MX-80, and the corresponding average basal spacings of the [Na-montmorillonite originating from Wyoming bentonite MX-80] samples reported in this study may partly be due to water contents and partly to the fact that only apparent \(\mathrm{d_{001}}\) values using Bragg’s law, without any profile fitting, were reported in their studies.
If
Kozaki et al. (2001)
should be used to support a claim about “interparticle porosity”, it
consequently has to be done in opposition to — not in conjunction
with —
Holmboe et al. (2012).
It would then also be appropriate for authors to provide arguments for
why they discard the conclusions of
Holmboe et al. (2012).4
Stacks in compacted bentonite make no geometrical sense
The literature is full of fancy figures of bentonite structure involving stacks. A typical example is found in Wu et al. (2018), and looks similar to this:
This illustration is part of a figure with the caption “Schematic representation of the different porosities in bentonite and the potential diffusion paths.”5 The regular rectangles in this picture illustrate stacks that each seems to contain five TOT-layers (I assume this throughout). Conveniently, these groups of five layers have the same length within each stack, while the length varies somewhat between stacks. This is a quite common feature in figures like this, but it is also common that all stacks are given the same length.
Another feature this illustration has in common with others is that the particles are ordered: we are always shown edges of the TOT-layers. I guess this is partly because a picture of a bunch of stacks seen from “the top” would be less interesting, but it also emphasizes the problem of representing the third dimension: figures like these are in practice figures of straight lines oriented in 2D, and the viewer is implicitly required to imagine a 3D-version of this two-dimensional representation.
A “realistic” stack picture
But, even as a 2D-representation, these figures are not representative
of what an actual configuration of stacks of TOT-layers looks like.
Individual TOT-layers have a distinct thickness of about 1 nm, but
varies widely in the other two dimensions.
Ploehn and Liu (2006)
analyzed the size distribution of Na-montmorillonite (“Cloisite
Na+”) using atomic force microscopy, and found an average aspect
ratio of 180 (square-root of basal area divided by thickness). A
representative single “TOT-line” drawn to scale is consequently
quite different from what is illustrated in in most stack-pictures,
and look like this (click on the figure to see it in full size)
In this figure, we have added “water layers” on each side of the TOT-layer (light red), with the water-to-solid volume ratio of 16. Neatly stacking five such units shows that the rectangles in the Wu et al. (2018)-figure should be transformed like this
But this is still not representative of what an assemblage of five
randomly picked TOT-layers would look like, because the size
distribution has a substantial variance. According to
Ploehn and Liu (2006), the
aspect ratio follows approximately a log-normal distribution. If we
draw five values from this distribution for the length of five
“TOT-lines”, and form assemblages, we end up with structures that
look like this:7
These are the kind of units that should fill the bentonite illustrations. They are quite irregularly shaped and are certainly not identical (this would be even more pronounced when considering the third dimension, and if the stacks contain more layers).
It is easy to see that it is impossible to construct a dense structure
with these building blocks, if they are allowed a random
orientation. The resulting structure rather looks something like this
Such a structure evidently has very low density, and are reminiscent of the gel structures suggested in e.g. Shalkevich et al. (2007) (see fig. 7 in that paper). This makes some sense, since the idea of stacks of TOT-layers (“tactoids”) originated from studies of low density structures, as discussed above.
Note that the structure in pictures like that in Wu et al. (2018) has a substantial density only because it is constructed with stacks with an unrealistic shape. But even in these types of pictures is the density not very high: with some rudimentary image analysis we conclude that the density in the above picture is only around 800 kg/m3. Also the figure from Navarro et al. (2017) above gives a density below 1000 kg/m3, although there it is explicitly stated that it is a representation of “highly compacted bentonite”.
The only manner in which the “realistic” building blocks can be
made to form a dense structure is to keep them in the same
orientation. The resulting structures then look e.g. like this
where we have color coded each stack, to remind ourselves that these
units are supposed to be fundamental.
Just looking at this structure of a “stack of stacks” should make it clear how flawed the idea is of stacks as fundamental structural units in compacted bentonite (note also how unrepresentative the stack-pictures found in the literature are). One of many questions that immediately arises is e.g. why on earth the tiny gaps between stacks (indicated by arrows) should remain. This brings us to the next argument against stacks as fundamental units for compacted water saturated bentonite:
What is supposed to keep stacks together?
Compacted bentonite of interest e.g. for sealing in radioactive waste repositories exerts swelling pressure of several MPa when in contact with external water. This osmotic pressure is a consequence of the presence of the mobile exchangeable cations in montmorillonite. Each “realistic” unit that we have imagined above is thus required to be at a huge elevated pressure, and the individual TOT-layers have a strong driving force to separate. And, unless a mechanism is provided for why such a separation is impossible, this is of course what we expect to happen! As far as I am aware, such a separation inhibiting mechanism has never been suggested in any publication that promotes the stack concept in compacted bentonite. To get a feel for the absurdity of this issue, let’s take a new look at the figure from Navarro et al. (2017)
Assuming that this system is in equilibrium with an external water
reservoir at zero pressure (i.e. atmospheric absolute pressure), the
pressure in the compartment labeled “intra-aggregate space” is also
close to zero. On the other hand, in the “stacks” located just a few
nm away, the pressure is certainly above 10 MPa in many places! A
structure like this is obviously not in mechanical equilibrium! (To use
the term “obvious” here feels like such an understatement.)
Implications
To sum up what we have discussed so far, the following picture
emerges. The bentonite literature is packed with descriptions of
compacted water saturated bentonite as built up of stacks as
fundamental units. These descriptions are so commonplace that they
often are not supported by references. But when they are, it seems
that the entire notion is based on misconceptions. In particular,
structures identified in low density systems (suspensions, gels) have
been carried over, without reflection, to descriptions of compacted
bentonite. Moreover, all figures illustrating the stack concept are
based on inadequate representations of what an arbitrary assemblage of
TOT-layers arranged in this way actually would look like. With a
“realistic” representation it quickly becomes obvious that it makes
little sense to base a fundamental unit in compacted systems on the
stack concept.
My impression is that this flawed stack concept underlies the entire
contemporary mainstream view of compacted bentonite, as e.g. expressed
by
Wu et al. (2018):
A widely accepted view is that the total porosity of bentonite
consists of \(\epsilon_ {ip}\) and \(\epsilon_ {il}\) (Tachi and
Yotsuji, 2014; Tournassat and Appelo, 2011; Van Loon et al., 2007).
\(\epsilon_ {ip}\) is a porosity related to the space between the
bentonite particles and/or between the other grains of minerals
present in bentonite. It can further be subdivided into
\(\epsilon_ {ddl}\) and \(\epsilon_ {free}\). The diffuse double layer,
which forms in the transition zone from the mineral surface to the
free water space, contains water, cations and a minor amount of
anions. The charge at the negative outer surface of the
montmorillonite is neutralized by an excess of cations. The free
water space contains a charge-balanced aqueous solution of cations
and anions. \(\epsilon_ {il}\) represents the space between TOT-layers
in montmorillonite particles exhibiting negatively charged
surfaces. Due to anion exclusion effect, anions are excluded from
the interlayer space, but water and cations are present.
This view can be summarized as:
The fundamental building blocks are stacks of TOT-layers
(“particles”, “aggregates”, “tactoids”, “grains”…)
Electric double layers are present only on external
surfaces of the stacks.
Far away from external surfaces — in the “inter-particle” or
“inter-aggregate” pores — the diffuse layers merge with a bulk
water solution
Interlayer pores are defined as being internal to the stacks,
and are postulated to be fundamentally different from the external
diffuse layers; they play by a different set of rules.
I don’t understand how authors can get away with promoting this
conceptual view without supplying reasonable arguments for all of its
assumptions8 — and with such a
complex structure, there are a lot of assumptions.
As already discussed, the geometrical implications of the suggested structure do not hold up to scrutiny. Likewise, there are many argumentsagainst the presence of substantial amounts of bulk water in compacted bentonite, including the pressure consideration above. But let’s also take a look at what is stated about “interlayers” and how these are distinguished from electric double layers (I will use quotation marks in the following, and write “interlayers” when specifically referring to pores defined as internal to stacks).
“Interlayers”
“Interlayers” are often postulated to be completely devoid of anions. We discussed this assumption in more depth in a previous blog post, where we discovered that the only references supplied when making this postulate are based on the Poisson-Boltzmann equation. But this is inadequate, since the Poisson-Boltzmann equation does describe diffuse layers, and predicts anions everywhere.
By requiring anion-free “interlayers”, authors actually claim that the physico-chemistry of “interlayers” is somehow qualitatively different from that of “external surfaces”, although these compartments have the exact same constitution (charged TOT-layer surface + ions + water). But an explanation for why this should be the case is never provided, nor is any argument given for why diffuse layer concepts are not supposed to apply to “interlayers”.9 This issue becomes even more absurd given the strong empirical evidence for that anions actually do reside in interlayers.
The treatment of anions is not the only ad hoc description of “interlayers”. It also seems close to mandatory to describe them as having a maximum extension, and as having an extension independently parameterized by sample density. E.g. the influential models for Na-bentonite of Bourg et al. (2006) and Tournassat and Appelo (2011) both rely on the idea that “interlayers” swell out to a certain volume that is smaller than the total pore volume, but that still depends on density.
In e.g. Bourg et al. (2006), the fraction of “interlayer” pores remains essentially constant at ~78%, as density decreases from 1.57 g/cm3 to 1.27 g/cm3, while the “interlayers” transform from having 2 monolayers of water (2WL) to having 3 monolayers (3WL). This is a very strange behavior: “interlayers” are acknowledged as having a swelling potential (2WL expands to 3WL), but do, for some reason, not affect 22% of the pore volume! Although such a behavior strongly deviates from what we expect if “interlayers” are treated with conventional diffuse layer concepts, no mechanism is provided.
Another type of macabre consequence of defining “interlayer” pores as internal to stacks is that a completely homogeneous system is described has having no interlayer pores (because it has no stacks). E.g. Tournassat and Appelo (2011) write (\(n_c\) is the number of TOT-layers in a stack)
[…] the number of stacks in the \(c\)-direction has considerable influence on the interlayer porosity, with interlayer porosity increasing with \(n_c\) and reaching the maximum when \(n_c \approx 25\). The interlayer porosity halves with \(n_c\) when \(n_c\) is smaller than 3, and becomes zero for \(n_c = 1\).10
It is not acceptable that using the term interlayer should require
accepting stacks as fundamental units. But the usage of the term as
being internal to stacks is so widespread in the contemporary
bentonite literature, that I fear it is difficult to even communicate
this issue. Nevertheless, I am certain that e.g.
Norrish (1954) does not
depend on the existence of stacks when using the term like this:
Fig. 7 shows the relationship between interlayer spacing and water
content for Na-montmorillonite. There is good agreement between the
experimental points and the theoretical line, showing that
interlayer swelling accounts for all, or almost all, of physical
swelling.
The stack view obstructs real discovery
A severe consequence of the conceptual view just discussed is that “stacking number” — the (average) number of TOT-layers that stacks are supposed to contain — has been established as fitting parameter in models that are clearly over-parameterized. An example of this is Tournassat and Appelo (2011), who write11
Our predictive model excludes anions from the interlayer space and
relates the interlayer porosity to the ionic strength and the
montmorillonite bulk dry density. This presentation offers a good
fit for measured anion accessible porosities in bentonites over a
wide range of conditions and is also in agreement with microscopic
observations.
But since anions do reside in interlayers,12 it would be better if the model didn’t fit: an over-parameterized or conceptually flawed model that fits data provides very little useful information.
A similar more recent example is Wu et al. (2018). In this work, a model based on the stack concept is successfully fitted both to data on \(\mathrm{ReO_4^-}\) diffusion in “GMZ” bentonite and to data on \(\mathrm{Cl^-}\) diffusion in “KWK” bentonite, by varying “stacking number” (among other parameters). Again, as the model assumes anion-free “interlayer” pores, a better outcome would be if it was not able to fit the data. Moreover, this paper focuses mainly on the ability of the model, while not at all emphasizing the fact that about ten (!) times more \(\mathrm{ReO_4^-}\) was measured in “GMZ” as compared with \(\mathrm{Cl^-}\) in “KWK”, at similar conditions in certain cases. The latter observation is quite puzzling and is, in my opinion, certainly worth deeper investigation (and I am fully convinced that it is not explained by differences in “stacking number”).
[3] Note that “sodium
dominated” in this context means ~20% or more.
[4] It may be noticed that Kozaki et al. (2001) see no X-ray diffraction peaks for low density samples:
The basal spacing of water-saturated
montmorillonite was determined by the XRD method. […] It was found
that a basal spacing of 1.88 nm, corresponding to the three-water
layer hydrate state […] was not observed before the dry density
reached 1.0 Mg/m3.
My interpretation of this observation is that the diffraction peak has
shifted to even lower reflection angles (in agreement with the
observations
of Holmboe
et al. (2012)), not registered by the equipment. The alternative
interpretation must otherwise be that “stacks” suddenly cease to
exist below 1.0 g/cm3. (Yet,
Kozaki et al. (2001)
continues to use a certain d-value in their analysis, also for densities
below 1.0 g/cm3.)
[5] I have discussed “diffusion
paths” in an
earlier blog post.
This illustration certainly fits that discussion.
[6] A water-to-solid volume ratio of 1 corresponds basically to
interlayers of three monolayers of water (3WL).
[7] To construct these units, I made the additional choice of placing each layer randomly in the horizontal direction, with the constraint that all layers should be confined within the range of the longest one in each unit.
[8] By “get away with” I mean “pass peer-review”, and by “don’t understand” I mean “understand”.
[10] A mathematical remark: if the interlayer porosity “halves with \(n_c\)” (what does that mean?) when \(n_c = 2\) (“smaller than 3”), it is impossible to simultaneously have zero interlayer porosity for \(n_c = 1\) (unless the interlayer porosity is zero for any \(n_c\)).
[11] I guess the word “presentation” here really should be “representation”?
[12] Note that one of the authors of this paper also claims in a later paper that anions do populate 3-waterlayer interlayers, in accordance with the Poisson-Boltzmann equation:
The agreement
between PB calculations and MD simulation predictions was somewhat
worse in the case of the \(\mathrm{Cl^-}\) concentration profiles than
in the case of the \(\mathrm{Na^+}\) profiles (Figure 3), perhaps
reflecting the poorer statistics for interlayer Cl concentrations
[…] Nevertheless, reasonable quantitative agreement was found
(Table 2).
Recently, we discussed reported equilibrium chloride concentrations in sodium dominated bentonite, and identified a need to assess the individual studies. As most data is obtained from through-diffusion experiments, we here take a general look at how anion equilibrium is a part of the through-diffusion set-up, and how we can use reported model parameters to extract the experimentally accessible equilibrium concentrations.
We define the experimentally accessible concentration of a chemical
species in a bentonite sample as
where \(n\) is the total amount of the
species,1 and \(m_{w}\) is
the total water mass in the
clay.2 It should be clear
that \(\bar{c}\), which we will refer to as the clay concentration, is
accessible without relying on any particular model concept.
An equilibrium concentration is defined as the corresponding clay concentration (i.e. \(\bar{c}\)) of a species when the clay is in equilibrium with an external solution with species concentration \(c^\mathrm{ext}\). A convenient way to express this equilibrium is in terms of the ratio \(\bar{c}/c^\mathrm{ext}\).
The through-diffusion set-up
A through-diffusion set-up consists of a (bentonite) sample sandwiched between a source and a target reservoir, as illustrated schematically here (for some arbitrary time):
The sample length is labeled \(L\), and we assume the sample to be
initially empty of the diffusing species. A test is started by adding
a suitable amount of the diffusing species to the source
reservoir. Diffusion through the bentonite is thereafter monitored by
recording the concentration evolution in the target
reservoir,3 giving
an estimation of the flux out of the sample (\(j^\mathrm{out}\)). The
clay concentration for anions is typically lower than the
corresponding concentration in the source reservoir.
Although a through-diffusion test is not in full equilibrium (by definition), local equilibrium prevails between clay and external solution4 at the interface to the source reservoir (\(x=0\)). Thus, even if the source concentration varies, we expect the ratio \(\bar{c}(0)/c^\mathrm{source}\) to stay constant during the course of the test.5
The effective porosity diffusion model
Our primary goal is to extract the concentration ratio \(\bar{c}(0)/c^\mathrm{source}\) from reported through-diffusion parameters. These parameters are in many anion studies specific to the “effective porosity” model, rather than being accessible directly from the experiments. We therefore need to examine this particular model.
The effective porosity model divides the pore space into a bulk water domain and a domain that is assumed inaccessible to anions. The porosity of the bulk water domain is often referred to as the “effective” or the “anion-accessible” porosity, and here we label it \(\epsilon_\mathrm{eff}\).
Anions are assumed to diffuse in the bulk water domain according to
Fick’s first law
where \(D_p\) is the pore diffusivity in the bulk water phase. This
relation is alternatively expressed as
\(j = -D_e \cdot \nabla c^\mathrm{bulk}\), which defines the effective
diffusivity \(D_e = \epsilon_\mathrm{eff} \cdot D_p\).
Diffusion is assumed to be the only mechanism altering the
concentration, leading to Fick’s second law
where \(\phi\) is the physical porosity of the sample. Since a bulk water concentration varies continuously across interfaces to external solutions, we have \(c^\mathrm{bulk}(0) = c^\mathrm{source}\) at the source reservoir, giving
This equation shows that the effective porosity parameter quantifies
the anion equilibrium concentration that we want to extract. That is
not to say that the model is valid (more on that later), but that we
can use eq. 4 to translate reported model parameters to an
experimentally accessible quantity.
In principle, we could finish the analysis here, and use eq. eq. 4 as our main result. But most researchers do not evaluate the effective porosity in the direct way suggested by this equation (they may not even measure \(\bar{c}\)). Instead, they evaluate \(\epsilon_\mathrm{eff}\) from a fitting procedure that also includes the diffusivity as a parameter. It is therefore fruitful to also include the transport aspects of the through-diffusion test in our analysis.
From closed-cell diffusion tests, we know that the clay concentration evolves according to Fick’s second law, both for many cations and anions. We will therefore take as an experimental fact that \(\bar{c}\) evolves according to
This equation defines the diffusion coefficient \(D_\mathrm{macr.}\), which should be understood as an empirical quantity.
Combining eqs. 3 and 2 shows that \(D_p\) governs the evolution of \(\bar{c}\) in the effective porosity model (if \(\epsilon_\mathrm{eff}/\phi\) can be considered a constant). A successful fit of the effective porosity model to experimental data thus provides an estimate of \(D_\mathrm{macr.}\) (cf. eq. 5), and we may write
With the additional assumption of constant reservoir concentrations,
eq. 2 has a relatively simple analytical solution, and the
corresponding outflux reads
where \(j^\mathrm{ss}\) is the steady-state flux. In steady-state,
\(c^\mathrm{bulk}\) is distributed linearly across the sample, and we
can express the gradient in eq. 1 using the reservoir
concentrations, giving
Treating \(j^\mathrm{ss}\) as an empirical parameter (it is certainly
accessible experimentally), and using eq. 6, we get another expression
for \(\epsilon_\mathrm{eff}\) in terms of experimentally accessible
quantities
This relation (together with eqs. 4 and 6) demonstrates that if we fit eq. 7 using \(D_p\) and \(j^\mathrm{ss}\) as fitting parameters, the equilibrium relation we seek is given by
This procedure may look almost magical, since any explicit reference to the effective porosity model has now disappeared; eq. 10 can be viewed as a relation involving only experimentally accessible quantities.
But the validity of eq. 10 reflects the empirical fact that the (steady-state) flux can be expressed using the gradient in \(\bar{c}\) and the physical porosity. The effective porosity model can be successfully fitted to anion through-diffusion data simply because it complies with this fact. Consequently, a successful fit does not validate the effective porosity concept, and essentially any description for which the flux can be expressed as \(j = -\phi\cdot D_p \cdot \nabla\bar{c}\) will be able to fit to the data.
We may thus consider a generic model for which eq. 5 is valid and for which a steady-state flux is related to the external concentration difference as
where \(\beta\) is an arbitrary constant. Fitting such a model, using \(\beta\) and \(D_p\) as parameters, will give an estimate of \(\bar{c}(0)/c^\mathrm{source}\) (\(=\beta / \phi\)).
Note that the system does not have to reach steady-state — eq. 11 only states how the model relates a steady-state flux to the reservoir concentrations. Moreover, the model being fitted is generally numerical (analytical solutions are rare), and may account for e.g. possible variation of concentrations in the reservoirs, or transport in the filters connecting the clay and the external solutions.
The effective porosity model emerges from this general description by interpreting \(\beta\) as quantifying the volume of a bulk water phase within the bentonite sample. But \(\beta\) can just as well be interpreted e.g. as an ion equilibrium coefficient (\(\phi\cdot \Xi = \beta\)), showing that this description also complies with the homogeneous mixture model.
Additional comments on the effective porosity model
The effective porosity model can usually be successfully fitted to anion through-diffusion data (that’s why it exists). The reason is not because the data behaves in a manner that is difficult to capture without assuming that anions are exclusively located in a bulk water domain, but simply because this model complies with eqs. 5 and 11. We have seen that also the homogeneous mixture model — which makes the very different choice of having no bulk water at all within the bentonite — will fit the data equally well: the two fitting exercises are equivalent, connected via the parameter identification \(\epsilon_\mathrm{eff} \leftrightarrow \phi\cdot\Xi\).
Perhaps even more remarkable is that authors frequently treat the effective porosity model as was it some version of the traditional diffusion-sorption model. This is often done by introducing a so-called rock capacity factor \(\alpha\) — which can take on the values \(\alpha = \phi + \rho\cdot K_d\) for cations, and \(\alpha = \epsilon_\mathrm{eff}\) for anions — and write \(D_e = \alpha D_a\), where \(D_a\) is the “apparent” diffusion coefficient. The reasoning seems to go something like this: since the parameter in the governing equation in one model can be written as \(D_e/\epsilon_\mathrm{eff}\), and as \(D_e/(\phi + \rho\cdot K_d)\) in the other, one can view \(\epsilon_\mathrm{eff}\) as being due to negative sorption (\(K_d < 0\)).
But such a mixing of completely different mechanisms (volume restriction vs. sorption) is just a parameter hack that throws most process understanding out the window! In particular, it hides the fact that the effective porosity and diffusion-sorption models are incompatible: their respective bulk water domains have different volumes. Furthermore, this lumping together of models has led to that anion diffusion coefficients routinely are reported as “apparent”, although they are not; the underlying model contains a pore diffusivity (eq. 2). As I have stated before, the term “apparent” is supposed to convey the meaning that what appears as pure diffusion is actually the combined result of diffusion, sorption, and immobilization. Sadly, in the bentonite literature, “apparent diffusivity” often means “actual diffusivity”.
Footnotes
[1] For anions, the total amount is relatively easy to measure by e.g. aqueous extraction. Cations, on the other hand, will stick to the clay, and need to be exchanged with some other type of cation (not initially present). In any case, the total amount of a species (\(n\)) can in principle be obtained experimentally, in an unambiguous manner.
[2] Another reasonable choice would be to divide by the total sample volume.
[3] If the test is designed as to have a significant change of the source concentration, it is a good idea to also measure the concentration evolution in this reservoir.
[5] Provided that the rest of the aqueous chemistry remains constant, which is not always the case. For instance, cation exchange may occur during the course of the test, if the set-up involves more than one type of cation, and there may be ongoing mineral dissolution.
In a
previous blog post,
we discussed how the diffusivity of simple
cations1
has a small, or even negligible, dependence on background
concentration (or, equivalently, on \(K_d\)), and how this observation
motivates modeling compacted bentonite as a homogeneous system,
containing only interlayer pores.
Despite the indisputable fact that “\(D_a\)”2 for simple ions does not depend much
on \(K_d\), the results have seldom been modeled using a homogeneous
bentonite model. Instead there are numerous attempts in the bentonite
literature to both measure and model a variation of “\(D_a\)” with
\(K_d\), usually with a conclusion (or implication) that “\(D_a\)”
depends significantly on \(K_d\). In this post we re-examine some of
these studies.
The claimed \(K_d\)-dependency is often “supported” by the so-called surface diffusion model. I have previously shown that this model is incorrect.3 Here we don’t concern ourselves with the inconsistencies, but just accept the resulting expression as the model to which authors claim to fit data. This model expression is
where \(D_p\) and \(D_s\) are individual domain diffusivities for bulk
water and surface regions, respectively, \(\rho\) is dry density, \(\phi\)
porosity, and \(K_d\), of course, is assumed to quantify the
distribution of ions between bulk water and surfaces as
\(s = K_d\cdot c^\mathrm{bulk}\), where \(s\) is the amount of ions on the
surface (per unit dry mass), and \(c^\mathrm{bulk}\) is the
corresponding bulk water concentration.
Muurinen et al. (1985)
Muurinen et al. (1985)
measured diffusivity in high density bentonite samples at various
background concentrations, using a type of closed-cell set-up. They
also measured corresponding values of \(K_d\) in batch “sorption”
tests. The results for cesium, in samples with density in the range
\(1870 \;\mathrm{kg/m^3}\) — \(2030 \;\mathrm{kg/m^3}\), are presented in
the article in a figure similar to this:
The markers show experimental data, and the solid curve shows the model (eq. 1) with \(D_p = 1.2 \cdot 10^{-10}\;\mathrm{m^2/s}\)4 and \(D_s = 4.3\cdot 10^{-13}\;\mathrm{m^2/s}\).
The published plot may give the impression of a systematic variation
of \(D_a\) for cesium, and that this variation is captured by the model.
But the data is plotted with a logarithmic y-axis, which certainly is
not motivated. Let’s see how the plot looks with a linear y-axis (we
keep the logarithmic x-axis, to clearly see the model variation).
Now the impression is quite different: this way of plotting reveals
that the experimental data only cover a part where the model does not
vary significantly. With the adopted range on the x-axis (as used in
the article) we actually don’t see the full variation of the model
curve. Extending the x-axis gives the full picture:
With the full model variation exposed, it is evident that the model fits the data only in a most superficial way. The model “fits” only because it has insignificant \(K_d\)-dependency in the covered range, in similarity with the measurements.
The defining feature of the model is that the diffusivity is supposed
to transition from one specific value at high \(K_d\), to a
significantly different value at low \(K_d\). As no such transition is
indicated in the data, the above “fit” does not validate the model.
Muurinen et al. (1985)
also measured diffusion of strontium in two samples of density
\(1740 \;\mathrm{kg/m^3}\). The figures below show the data and
corresponding model curve.
The left diagram is similar to how the data is presented in the article, while the right diagram utilizes a linear y-axis and shows the full model variation. The line shows the surface diffusion model with parameters \(D_p = 1.2 \cdot 10^{-10}\;\mathrm{m^2/s}\) and \(D_s = 8.8 \cdot 10^{-12}\;\mathrm{m^2/s}\). In this case it is clear even from the published plot that the experimental data shows no significant variation.
The only reasonable conclusion to make from the above data is that cesium and strontium diffusivity does not significantly depend on \(K_d\) (which implies a homogeneous system). This is actually also done in the article:
The apparent diffusivities of strontium and cesium do not change
much when the salt concentration used for the saturation of the
samples is changed and the sorption factors change. The surface
diffusion model agrees fairly well with the observed
diffusion-sorption behaviour.
I agree with the first sentence but not with the second. In my mind,
the two sentences contradict each other. From the above plots,
however, it is trivial to see that the surface diffusion model does
not agree (in any reasonable sense) with observations.
Eriksen et al. (1999)
Although
Muurinen et al. (1985)
concluded insignificant \(K_d\)-dependency on the diffusion coefficients
for strontium and cesium, researchers have continued throughout the
years to fit the surface diffusion model to experimental data on these
and other ions.
Eriksen et al. (1999) present old and new diffusion data for strontium and cesium (and sodium), fitted and plotted in the same way as in Muurinen et al. (1985). Here are the evaluated diffusivities for cesium plotted against evaluated \(K_d\), as presented in the article, and re-plotted in different ways with a linear y-scale:
The curve shows the surface diffusion model (eq. 1), with parameters \(D_p = 8 \cdot 10^{-10}\;\mathrm{m^2/s}\) and \(D_s = 6 \cdot 10^{-13}\;\mathrm{m^2/s}\). The points labeled “Eriksen 99” are original data obtained from through-diffusion tests on “MX-80” bentonite at dry density 1800 \(\mathrm{kg/m^3}\).5 The source for the data points labeled “Muurinen 94” is the PhD thesis of A. Muurinen.6
The upper left plot shows the data as presented in the article; again,
a logarithmic y-axis is used. In this case, a zoomed-in view with a
linear y-axis (upper right diagram) may still give the impression that
the data has a systematic variation that is captured by the model. But
viewing the whole range reveals that the model is fitted to data where
variation is negligible (bottom diagrams), just as in
Muurinen et al. (1985).
The model (line) has parameters \(D_p = 3 \cdot 10^{-10}\;\mathrm{m^2/s}\) and \(D_s = 1 \cdot 10^{-11}\;\mathrm{m^2/s}\), and the source for the data points labeled “Eriksen 84” is found here.
In this case, not even the diagram presented in the article (left)
seems to support the promoted model. This is also confirmed when
utilizing a linear y-axis, and showing the full model variation (right
diagram).
Eriksen et al. (1999)
conclude that strontium diffusivities are basically independent of
\(K_d\), but claim, in contrast to
Muurinen et al. (1985),
that cesium diffusivity depends significantly on \(K_d\):
[I]n the \(K_d\) interval 0.01 to 1 the apparent \(\mathrm{Cs}^+\)
diffusivity decreases by approximately one order of magnitude
whereas for \(\mathrm{Na}^+\) and \(\mathrm{Sr}^{2+}\) the apparent
diffusivity is virtually constant.
They also claim that the surface diffusion model fits the data:
\(D_\mathrm{a}\) curves for \(\mathrm{Cs}^+\) and \(\mathrm{Sr}^{2+}\), calculated using a Eq. (6) [eq. 1 here], are plotted in Fig. 4. As can be seen, good fits to experimental data were obtained […]
Note that the variation in the model for cesium is motivated by three data points with relatively high diffusivity and basically the same \(K_d \sim 0.05\;\mathrm{m^3/kg}\). It seems like the model has been fitted to these points, while the point at \(K_d \sim 0.02\;\mathrm{m^3/kg}\) has been mainly neglected. The resulting model has a huge bulk water diffusivity (\(D_p\)), which is about 7 times larger than in the corresponding fit in Muurinen et al. (1985), and only 2.5 times smaller than the diffusivity for cesium in pure water.
Note that, if you claim that the surface diffusion model fits in this
case, you implicitly claim that the observed variation — which still
is negligible on the scale of the full model variation — is
caused by the influence of this enormous (for a 1800
\(\mathrm{kg/m^3}\) sample) bulk pore water diffusivity; with a more
“reasonable” value for \(D_p\), the model no longer fits. There are
consequently valid reasons to doubt that the claimed \(K_d\) dependence
is real. We will return to this fit in the next section.
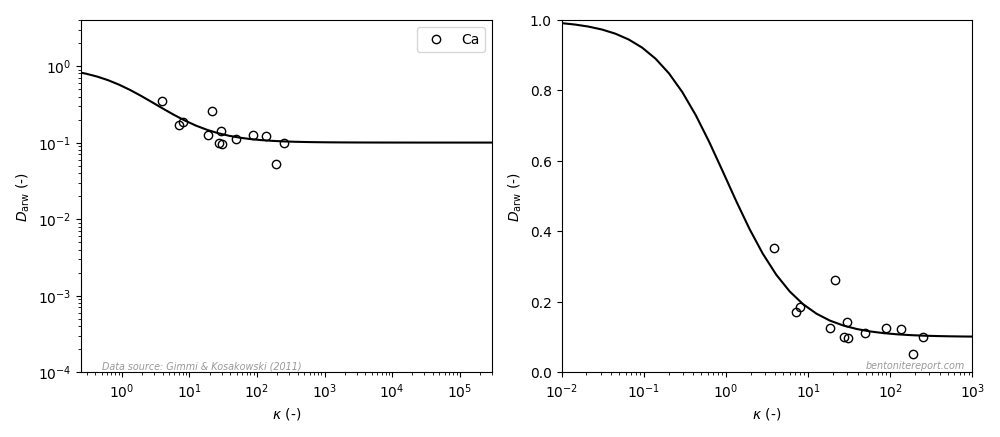
Gimmi & Kosakowski (2011)
We have now seen several examples of authors erroneously claiming (or
implying) that a surface diffusion model is valid, when the actual
data for “\(D_a\)” has no significant \(K_d\)-dependency. For reasons I
cannot get my head around, this flawed treatment is still in play.
Rather than identifying the obvious problem with the previously
presented fits, Gimmi and
Kosakowski (2011) instead extended the idea of expressing the
diffusivity as a function of \(K_d\) by using scaled, dimensionless
quantities
where \(D_0\) is the corresponding diffusivity in pure water and
\(\tau_w\) is the “tortuosity factor” for water in the system of
interest. This factor is simply the ratio between the water
diffusivity in the system of interest and the water diffusivity in
pure water (I have written about the problem with factors like this
here).
The idea — it seems — is that using \(D_\mathrm{arw}\) and \(\kappa\) as
variables should make it possible to directly compare the mobility of
a given species in systems differing in density, clay content, etc.
Even though it makes some sense that the diffusivity of a specific
species scales with the diffusivity of water in the same system, the
above procedure inevitably introduces more variation in the data —
both because an additional measured quantity (water diffusivity) is
involved when evaluating the scaled diffusivity, but also because
water diffusivity may depend differently on density as compared with
the diffusivity of the species under study.
Also Gimmi and Kosakowski
(2011) use the flawed surface diffusion model for analysis, and
their expression for \(D_\mathrm{arw}\) is
where \(\mu_s = D_s\tau_w/D_0\) is a “relative surface mobility”. This equation is obtained from eq. 1, by dividing by \(D_p\) and assuming \(D_p = D_0/\tau_w\).
Gimmi and Kosakowski (2011) fit eq. 3 to a large set of collected data, measured in various types of material, including bentonites, clay rocks, and clayey soils. This is their result for cesium7 (the model curve is eq. 3 with \(\mu_s = 0.031\)8)
Viewed as a whole, this data is more scattered as compared with the previous studies. This is reasonably an effect of the larger diversity of the samples, but also an effect of multiplying the “raw” diffusion coefficient with the factor \(\tau_w\) (eq. 2).
Just as in the previous studies we have looked at, the published plot (similar to the left diagram) may give the impression of a systematic variation of the diffusivity with \(K_d\) (it contains partly the same data). But just as before, a linear y-axis (right diagram) reveals that the model is fitted only to data where variation is negligible.
Note that the three data points that contributed to the majority of the variation in the fitted model in Eriksen et al. (1999) here appear as outliers.9 The variation with \(K_d\) for cesium claimed in that study is thus invalidated by this larger data set.
As we have noted already, the only reasonable conclusion to draw from this data is that there is no systematic \(K_d\)-dependency on diffusivity of cesium or strontium, and that it does not — in any reasonable sense — fit the surface diffusion model. Yet, also Gimmi and Kosakowski (2011) imply that the surface diffusion is valid:
The data presented here show a general agreement with a simple
surface diffusion model, especially when considering the large
errors associated with the \(D_\mathrm{erw}\) and \(D_\mathrm{arw}\).
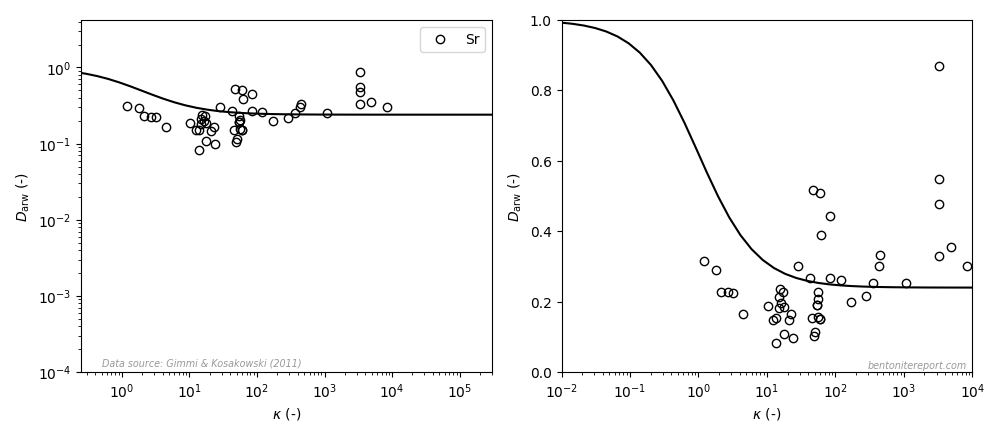
This paper, however, contains an even worse “fit” to strontium data, as compared to the earlier studies (the left diagram is similar to the how it is presented in the article, the right diagram uses a linear y-axis; the line is eq. 3 with \(\mu_s = 0.24\)8):
This data does not suggest a variation in accordance with the adopted model even when plotted in a log-log diagram. With a linear y-axis, the dependence rather seems to be the opposite: \(D_\mathrm{arw}\) appears to increase with \(\kappa\). However, I suspect that this is a not a “real” dependence, but rather an effect of trying to construct a “relative” diffusivity; note that while \(\kappa\) spans four orders of magnitude, \(D_\mathrm{arw}\) scatters only by a factor of 5 or 6. Nevertheless, how this data can be claimed to show “general agreement” with the surface diffusion model is a mystery to me.
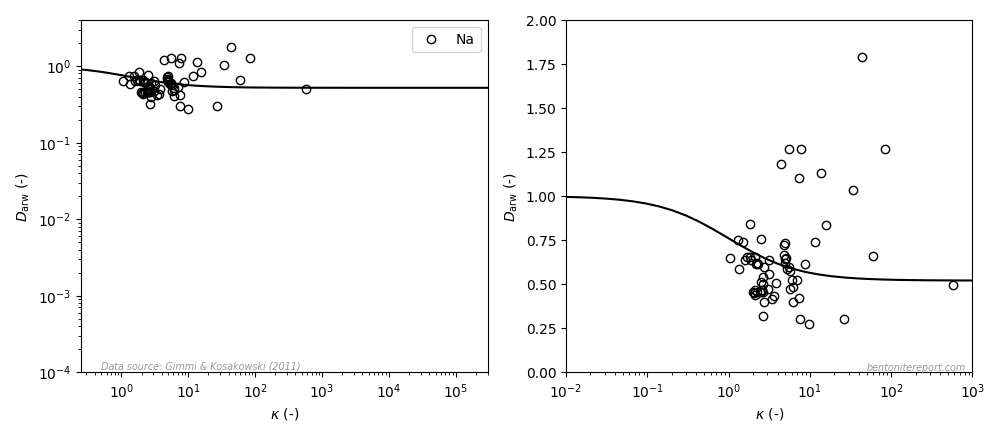
The view is similar for sodium (the left diagram is similar to the how it is presented in the article, the right diagram uses a linear y-axis; the line is eq. 3 with \(\mu_s=0.52\)8):
Even if the model in this case only displays minor variation, it can
hardly be claimed to fit the data: again, the data suggests a
diffusivity that increases with \(\kappa\). But a significant amount of
these data points have \(D_\mathrm{arw} > 1\), which is not likely to be
true, as it indicates that the relative mobility for sodium is larger
than for water. Consequently, the major contribution of the variation
seen in this data is most probably noise.
Gimmi and Kosakowski (2011) also examined diffusivity for calcium, and the data looks like this (the left diagram is similar to the how it is presented in the article, the right diagram uses a linear y-axis; the line is eq. 3 with \(\mu_s=0.1\)8):
Here it looks like the data, to some extent, behaves in accordance with the model also when plotted with linear y-axis covering the full model variation. However, there are significantly less data reported for calcium (as compared with cesium, strontium, and sodium) and the model variation is supported only by a few data points10. I therefore put my bet on that if calcium diffusivity is studied in more detail, the dependence suggested by the above plot will turn out to be spurious.11
Some thoughts
I am more than convinced that the only reasonable starting point for modeling saturated bentonite is a homogeneous description. I had nevertheless expected to at least have to come up with an argument against the multi-porous view put forward in the considered publications (and in many others). I am therefore quite surprised to find that this argument is already provided by the data in the very same publications (and even by the statements, sometimes): there is nothing in the data here reviewed that seriously suggests that cation diffusion is influenced by a heterogeneous pore structure.
Still, the unsupported idea that cations in compacted bentonite are supposed to diffuse in two (or more) different types of water domains has evidently propagated through the scientific literature for decades, and a multi-porous view is mainstream in modern bentonite research. It is difficult to not feel disheartened when faced with this situation. What would it take for researchers to begin scrutinize their assumptions? Is nobody interested in the topics we are supposed to study?
[2] I use quotation marks to indicate that \(D_a\) is a parameter in the traditional diffusion-sorption model, a model not valid for compacted bentonite. Still, this parameter is often reported as if it was a directly measured quantity.
[4] The article states
\(\epsilon D_p = 3.5\cdot 10^{-11}\; \mathrm{m^2/s}\), where
\(\epsilon\) is the porosity.
\(D_p = 1.2\cdot 10^{-10} \; \mathrm{m^2/s}\) corresponds to
\(\epsilon = 0.29\).
[5] In this study, both \(K_d\) and \(D_a\) were evaluated by fitting the traditional diffusion-sorption model to concentration measurements.
[6] I have had no access to this document, and I have not verified e.g. sample density (this data set is different from that presented in the previous section). Instead, I have read these values from the diagram in Eriksen et al. (1999).
[7] They actually divide their cesium data into two categories, which show quite different mobility. The data shown here — which includes bentonite samples — is for systems categorized as being “non-illite” or having Cs concentration above “trace”.
[8] According to the article table, the fitted values for \(\mu_s\) are 0.52 (Na), 0.39 (Sr), 0.087 (Ca), and 0.015 (Cs). The plotted lines, however, appear to instead use what is listed as “mean \(\mu_s\)”. Here, I have used these \(\mu_s\)-values: 0.52 (Na), 0.24 (Sr), 0.1 (Ca), and 0.031 (Cs).
[11] It would also be more than amazing if it turns out — after it is verified that Cs, Na, and (especially) Sr show no significant \(K_d\) dependence — that Ca diffusivity actually varies in accordance with the flawed surface-diffusion model!
We can directly apply the homogeneous mixture model for bentonite to isolated systems — e.g. closed-cell diffusion tests — as discussed previously. For systems involving external solutions we must also handle the chemical equilibrium at solution/bentonite interfaces.
I have presented a framework for calculating the chemical equilibrium between an external solution and a bentonite component in the homogeneous mixture model here. In this post I will discuss and illustrate some aspects of that work.
Overview
We assume a homogeneous bentonite domain in contact with an external solution, with the clay particles prevented from crossing the domain interface. For real systems, this corresponds to the frequently encountered set-up with bentonite confined in a sample holder by means of e.g. a metal filter. From the assumptions of the homogeneous model — that all ions are mobile and allowed to cross the domain interface — it follows that the type of equilibrium to consider is the famous Donnan equilibrium. I have discussed the Donnan effect and its relevance for bentonite quite extensively here.
Since the adopted model assumes a homogeneous bentonite domain, the
only region where Donnan equilibrium comes into play is at the
interface between the bentonite and the external solution. This is
quite different from how
Donnan equilibrium
calculations are implemented in many multi-porous models,
where the equilibrium is internal to the clay — between assumed
“macro” and “micro” compartments of the pore structure. The need
for performing Donnan equilibrium calculations is thus
minimized in the homogeneous mixture model (as mentioned,
isolated systems require no such calculations). Note also that the
semi-permeable mechanism in multi-porous models is required to act on
the pore-scale. I have never seen any description or explanation how
such a mechanism is supposed to work.1 In the
homogeneous mixture model, on the other hand, the semi-permeable
interface corresponds directly to a macroscopic and experimentally
well-defined component: the confining filter.
The problem to be solved can be illustrated like this
The aim is to relate the set of species concentrations in the external solution (\(\{c_i^\mathrm{ext}\}\)) to those in the clay domain (\(\{c_i^\mathrm{int}\}\)) when the system is in equilibrium. This is done by applying the standard approach to Donnan equilibrium, as found in textbooks on the subject. If there is anything “radical” about this framework, it is thus not in the way Donnan equilibrium is implemented, but rather in treating bentonite as a single phase: this approach is formally equivalent to assuming the bentonite to be an aqueous solution.
Chemical equilibrium
I prefer to formulate the Donnan equilibrium framework in a way that
separates effects due to difference in the local chemical environment
from effects due to differences in electrostatic potential between the
two compartments. An important reason for focusing on this separation
is that the local environment affects the chemistry under all
circumstances, while the (relative) value of the electrostatic
potential only is relevant when bentonite is contacted with an
external solution. We therefore express the chemical equilibrium as
This formula is achieved by setting the electro-chemical potential equal for each species in the two compartments. Here \(\gamma_i\) denotes the activity coefficient for species \(i\), and \(\psi^*\) is the
electrostatic potential difference between the compartments, which we refer to as the Donnan potential.
I find it convenient to rewrite this expression using some fancy Greek letters
Here I call \(\Xi_i = c_i^\mathrm{int}/c_i^\mathrm{ext}\) the ion equilibrium coefficient for species \(i\). This quantity expresses the essence of ion equilibrium in the homogeneous mixture model, and
will appear in many places in the analysis. \(\Xi_i\) has two factors:
\(\Gamma_i = \gamma_i^\mathrm{ext}/\gamma_i^\mathrm{int}\) expresses the chemical aspect of the equilibrium: when \(\Gamma_i\) is large (\(>1\)), the species has a chemical preference for residing in the interlayer pores, and when \(\Gamma_i\) is small (\(<1\)), the species has a preference for the external solution. In general, \(\Gamma_i\) for any specific species \(i\) is a function of all species concentrations in the system.
\(f_D^{-z_i}\), where \(f_D = e^{\frac{F\psi^\star}{RT}}\) is a dimensionless transformation of the Donnan potential (this is basically the Nernst equation), which we here call the Donnan factor. \(f_D\) expresses the electrostatic aspect of the equilibrium, and is the same for all species. The effect on \(\Xi_i\), however, is different for species of different charge number, because of the exponent \(-z_i\) in the full expression.
I want to emphasize that eqs. 1 and 2 express the exact same thing: chemical equilibrium between the two compartments.
Illustrations
To get a feel for the quantity \(\Xi\), here is a
hopefully useful animation
It may also be helpful to see the influence of \(f_D\) on the equilibrium. Since the Donnan potential is negative, \(f_D\) is less than unity and typical values in relevant bentonite systems is \(f_D \sim\) 0.01 — 0.4. Due to the exponent \(-z_i\) in eq. 2, this influence on the equilibrium looks quite different for species with different valency. For mono- and di-valent cations, the behavior looks like this (here is put \(\Gamma = 1\) for both species)
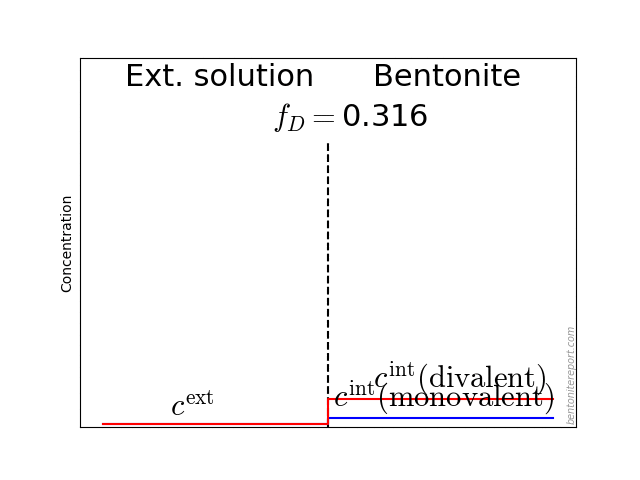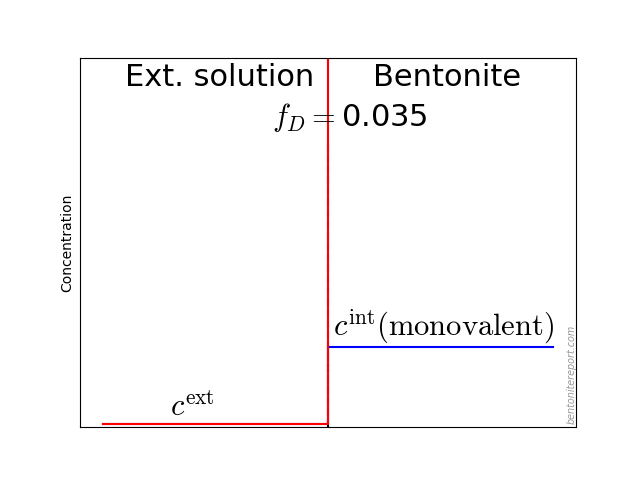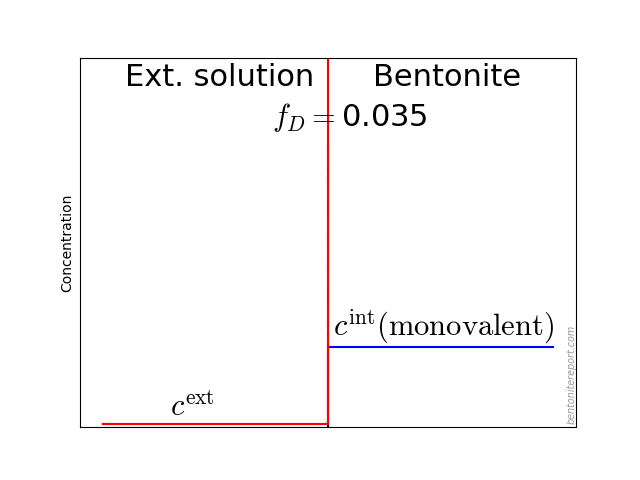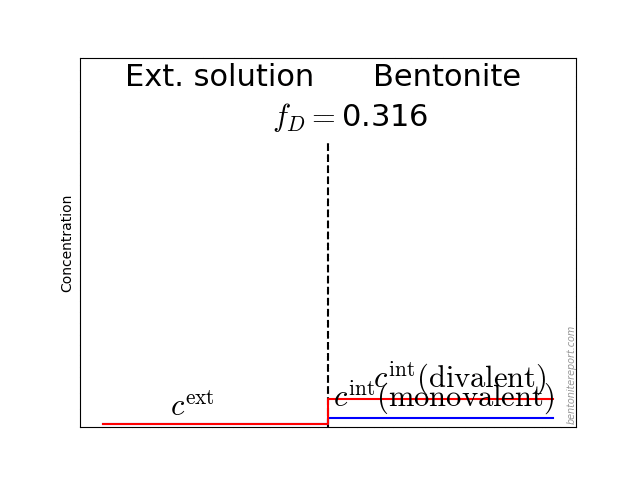
The typical behavior for cations is that the internal concentration is much larger than the corresponding external concentration (at \(f_D = 0.01\) in the above animation, the internal concentration for the di-valent cation is enhanced by a factor \(\Xi = 10 000\)!). For anions, the internal concentration is instead lower than the external concentration,2 as shown here (\(\Gamma = 1\) for both species)
Equation for \(f_D\)
For a complete description, we need an equation for calculating \(f_D\). This is derived by requiring charge neutrality in the two compartments and looks like
is the structural charge present in the clay (i.e. negative
montmorillonite layer charge) expressed as a monovalent interlayer
concentration. Here \(CEC\) is the cation exchange capacity of the clay
component, \(w\) the water-to-solid mass ratio,3 and \(F\) is the Faraday constant.
The way eq. 3 is formulated implies that the external concentrations should be used as input to the calculation. This is typically the case as the external concentrations are under experimental control.
In typical geochemical systems it is required to account for aqueous
species with valency at least in the range -2 — +2 (e.g.
\(\mathrm{Ca}^{2+}\), \(\mathrm{Na}^{+}\), \(\mathrm{Cl}^{-}\),
\(\mathrm{SO_4}^{2-}\)), which implies that the equation for calculating
\(f_D\) is generally a polynomial equation of degree four or higher.
An important special case is the 1:1 system — e.g. pure Na-montmorillonite contacted with a NaCl solution — which has an equation for \(f_D\) of only degree two, and thus has a relatively simple analytical solution
With the machinery in place for calculating the Donnan potential, here
is an animation demonstrating the response in internal sodium and chloride
concentrations as the external NaCl concentration is varied. In this
calculation \(c_{IL} = 2\) M, and
\(\Gamma_\mathrm{Na} = \Gamma_\mathrm{Cl} = 1\)
Comment on through-diffusion
To me, the last illustration makes it absolutely clear that Donnan equilibrium and the homogeneous mixture model provide the correct principal explanation for e.g. the behavior of tracer ions in through-diffusion tests. If you choose to relate the flux in through-diffusion tests to the external concentration difference — which is basically done in all published studies, via the parameter \(D_e\) — you will evaluate large “diffusivities” for cations and small “diffusivities” for anions. These “diffusivities” will, moreover, have the opposite dependence on background concentration: the cation flux diverges in the low background concentration limit,4 while the anion flux approaches zero.
But this behavior is seen to be caused by differently induced
internal concentration gradients. If fluxes are related to
these gradients — which they of course should, if you strive for an
actual Fickian description — you find that the diffusivities are no
different from what is evaluated in closed-cell tests. Relating the
steady-state flux to the external concentration difference in the
homogeneous mixture model gives (assuming zero tracer concentration on
the outflow side)
where \(c^\mathrm{source}\) denotes the tracer concentration in the external solution on the inflow side, \(\phi\) is the porosity, \(D_c\) is the pore diffusivity in the interlayer domain, and \(L\) is the length of the bentonite sample. From the above equation can directly be identified
When I first published on Donnan equilibrium in bentonite, I was a bit confused and singled out the term “Donnan equilibrium” to refer to anions only, while calling the corresponding cation equilibrium “ion-exchange equilibrium”. To refer to “both” types of equilibrium we used the term “ion equilibrium”.6 Of course, Donnan equilibrium applies to ions of any charge and, being better informed, I should have used a more stringent terminology. In later publications I have tried to make amends by pointing out that the process of cation exchange is part of the establishment of Donnan equilibrium.
Being new to the Donnan equilibrium world, I also invented some of my own nomenclature and symbols: e.g. I named the ratio between internal and external concentration the ion equilibrium coefficient (\(\Xi\)). Conventionally, if I now have understood correctly, this concentration ratio is referred to as the “Donnan ratio”, and is usually labeled \(r\) (although I’ve also seen \(K\)).
But the term “Donnan ratio” seems to be used slightly differently in different contexts, e.g. defined either as \(c^\mathrm{int}/c^\mathrm{ext}\) or as \(c^\mathrm{ext}/c^\mathrm{int}\), and is sometimes related more directly to the Donnan potential (if no distinction is made between activities and concentrations, we can write \(f_D^{-z_i} = c_i^\mathrm{int}/c_i^\mathrm{ext}\)). I therefore will continue to use the term “ion equilibrium coefficient” — with label \(\Xi\) — in the context of bentonite systems. This usage has also been picked up in some otherclay publications. The ion equilibrium coefficient should be understood as strictly defined as \(\Xi = c^\mathrm{int}/c^\mathrm{ext}\) for any species, and never to define, or being defined by, the Donnan potential.
To emphasize the difference between effects due to the presence of a
Donnan potential and effects due to different local chemical
environments, I will refer to \(f_D\) as the
Donnan factor. (This term
does not
seem to be used conventionally for any other quantity,
although there are examples where it is used as a synonym for Donnan
ratio.)
Finally, as in any other approach, the current framework requires a
description for the activity coefficients. For activity coefficients
in the external solution, there are
quitea numberof
models already available. For the interlayer, modeling — and
measuring! — activities is an open research area (at least I hope
that this research area is open).
[2] This does not have to be the case in principle, if \(\Gamma\) for the anion is large, at the same time as the external concentration is not too low.
[3] Hence, it is
implied that we use concentration units based on water mass
(molality).
[5] Speaking of “sorption”, we have noted before that this term nowadays is used to mean any type of uptake between bulk water and some other domain (where the species may or may not be immobile). In this sense, there is “sorption” in the homogeneous mixture model (for both cations and anions), but only at interfaces to external solutions. It thus translates to a boundary condition, rather than being part of the transport dynamics within the clay (which makes life much simpler from a numeric perspective). Update (220622): The homogeneous mixture model is extended to deal with ions that truly sorbs here.
Consider this basic experiment: contact a water saturated sample of compacted pure Na-montmorillonite, with dry mass 10 g and cation exchange capacity 1 meq/g, with an external solution of 100 ml 0.1 M KCl. Although such an experiment has never been reported1, I’m convinced that all agree that the outcome would be similar to what is illustrated in this animation.
Potassium diffuses in, and sodium diffuses out of the sample until equilibrium is established. At equilibrium also a minor amount of chloride is found in the sample. The indicated concentration levels are chosen to correspond roughly to results from from similar type of experiments.2
Although results like these are quite unambiguous, the way they are described and modeled in the bentonite3 literature is, in my opinion, quite a mess. You may find one or several of the following terms used to describe the processes
Cation exchange
Sorption/Desorptioṇ
Anion exclusion
Accessible porosity
Surface complexation
Donnan equilibrium
Donnan exclusion
Donnan porosity/volume
Stern layer
Electric double layer
Diffuse double layer
Triple layer
Poisson-Boltzmann
Gouy-Chapman
Ion equilibrium
…
In this blog post I argue for that the primary mechanism at play is
Donnan equilibrium, and that most of the above terms can be
interpreted in terms of this type of equilibrium, while some of the
others do not apply.
But I would like to push for that “Donnan equilibrium” primarily
should be the name of an observable
effect, and that it applies equally to both anions and
cations. This effect — which was
hypothesized by Gibbs already in the 1870s — relies basically only
on two things:
An electrolytic system, i.e. the presence of charged aqueous
species (ions).
The presence of a semi-permeable component that is permeable to
some of the charges, but does not allow for the passage of at least
one type of charge.
In equilibrated systems fulfilling these requirements it is — to use Donnan’s own words — “thermodynamically necessary” that the permeant ions distribute unequally across the semi-permeable component. This phenomenon — unequal ion distributions on the different sides of the semi-permeable component — should, in my opinion, be the central meaning of the term “Donnan equilibrium”.
The first publication of Donnan on the effect actually concerned osmotic pressure response, in systems of Congo Red separated from solutions of sodium chloride and sodium hydroxide. The same year (1911) he also published the ionic equilibrium equations for some specific systems.5 In particular he considered the equilibrium of NaCl initially separated from NaR, where R is an impermeant anion (e.g. that of Congo Red), leading to the famous relation (“int” denotes the solution containing R)
Unfortunately, this relation alone (or relations derived from it) is often what the term “Donnan” is associated with in today’s clay research literature, with the implication that systems not obeying it are not Donnan systems. But the above relation assumes ideal conditions and complete ionization of the salts — issues Donnan persistently seems to have grappled with. In a review on the effect he writes
The exact equations can, however, be stated only in terms of the chemical potentials of Willard Gibbs, or of the ion activities or ionic activity-coefficients of G. N. Lewis. Indeed an accurate experimental study of the equilibria produced by ionically semi-permeable membranes may prove to be of value in the investigation of ionic activity coefficients.
It must therefore be understood that, if in the following pages ionic concentrations and not ionic activities are used, this is done in order to present a simple, though only approximate, statement of the fundamental relationships.
The issue of (the degree of) ionization was explicitly addressed in publications following the 1911 article; Donnan & Allmand (1914) motivated their investigations of the \(\mathrm{KCl/K_4Fe(CN)_6}\) system by that “it was deemed advisable to test the relation when using a better defined, non-dialysable anion than that of Congo-red”, and the study of the Na/K equilibrium in Donnan & Garner (1919) used ferrocyanide solutions on both sides of the membrane in an attempt to overcome the difficulty of the “uncertainty as to the manner of ionisation of potassium ferrocyanide” (and thus for the simplified equations to apply).
I mean that since non-ideality and ion association are general issues when treating salt solutions, it does not make much sense to use the term “Donnan equilibrium” only when some particular equation applies; as long as the mechanism for the observed behavior is that some charges diffuse through a semi-permeable component, while some others don’t, the effect should be termed Donnan equilibrium.
Donnan equilibrium in gels, soils and clays
After Donnan’s original publications in 1911, the effect was soon recognized in colloidal systems. Procter & Wilson (1916) used Donnan’s equations to analyze the swelling of gelatin jelly immersed in hydrochloric acid. In this case chloride is the charge compensating ion, allowed to move between the phases, while the immobile charge is positive charges on the gelatin network. Thus, no semi-permeable membrane is necessary for the effect; alternatively one could say that the gel constitutes its own semi-permeable component. The Donnan equilibrium in protein solutions was further and extensively investigated by Loeb.
As far as I am aware, Mattson was first to identify the Donnan effect in “soil” suspensions,6 attributing e.g. “negative adsorption” of chloride as a consequence of Donnan equilibrium, and explicitly referencing the works of Procter and Loeb. Mattson describes the suspension in terms of electric double layers with a diffuse “atmosphere of cations” surrounding the “micelle” (the soil particle), and refers to Donnan equilibrium as the distribution of an electrolyte between the “micellar” and the “inter-micellar” solutions. Oddly,7 he uses Donnan’s original framework (e.g. eq. 1) to quantify the equilibrium, although the electrostatic potential and the ion concentrations varies significantly in the investigated systems. A more appropriate treatment would thus be to use e.g. the Gouy-Chapman description for the ion distribution near a charged plane surface (which he refers to!).
Instead, Schofield (1947) analyzed Mattson’s data using this approach. He also comments on its (the Gouy-Chapman model) range of validity
… [T]he equation is applicable to cases in which the distance
between opposing surfaces considerably exceeds the distance between
neighboring point charges on the surfaces; for there will then be a
range of electrolyte concentrations over which the radius of the
ionic atmosphere is less than the former and greater than the
latter. In Mattson’s measurements on bentonite suspension, these
distances are roughly 500 A. and 10 A. respectively, so there is an
ample margin.
He continues to comment on the validity of Donnan’s original equations
When the distance ratio has narrowed to unity, it is to be expected
that the system will conform to the equation of the Donnan membrane
equilibrium. This equation fits closely the measurements of Procter
on gelatine swollen in dilute hydrochloric acid. […] In a
bentonite suspension the charges are so far from being evenly
distributed that the Donnan equation is not even approximately
obeyed.
From these statements it should be clear that the general behavior (cation exchange, salt exclusion) of ions in bentonite equilibrated with an external solution is due to the Donnan effect.8 The appropriate theoretical treatment of this effect differs, however, depending on details of the investigated system. To argue whether or not e.g. the Gouy-Chapman description should be classified as a “Donnan” approach is purely semantic.
It is also clear that in the case of compacted bentonite the distance ratio is narrowed to unity — the typical interlayer distance is 1 nm, which also is the typical distance between structural charges in the montmorillonite particles. It is thus expected that Donnan’s original treatment may work for such systems (adjusted for non-ideality), while the Gouy-Chapman description is not valid.9
The message I am trying to convey is neatly presented in Overbeek (1956) — a text I highly recommend for further information. Overbeek distinguishes between “classical” (Donnan’s original) and “new” (accounting for variations in potential etc.) treatments of Donnan equilibrium, and says the following about dense systems
If the particles come very close together the potential drop between
[surface and interlayer midpoint] becomes smaller and smaller as
illustrated in Fig. 4. This means that the local concentrations of
ions are not very variable and that we are again back at the
classical Donnan situation, where distribution of ions, osmotic
pressure and Donnan potential are simply given by the elementary
equations as treated in section 2. It is remarkable that the new
treatment of the Donnan effects may deviate strongly from the
classical treatment when the colloid concentration is low, but not
when it is high.
It thus seems plausible that Donnan equilibrium in compacted bentonite can be treated using Donnan’s original equations. But — as interlayer pores are a quite extreme chemical environment — substantial non-ideal behavior may be expected. Treating such behavior is a large challenge for chemical modeling of compacted bentonite, but can not be avoided, since interlayers dominate the pore structure.
Cation exchange is Donnan equilibration
The term “Donnan” in modern bentonite literature is, as mentioned,
quite heavily associated with the fate of anions interacting with
bentonite. In contrast, cations are often described as being
“sorbed” onto the “solids”. This sorption is usually separated
into two categories: cation exchange and surface complexation.
Surface complexation reactions are typically described using
“surface sites”, and
are usually written
something like
this (exemplified with sodium sorption)
where the “surface site” is labeled \(\equiv \mathrm{S}^-\)
Cation exchange is also typically writtenintermsof“sites”, but requires the exchange of ions (duh!), like this (here exemplified for calcium/sodium exchange)
Underlying these modeling approaches and descriptions is the (sometimes implicit) idea that exchanged ions are immobile, which clearly has motivated e.g. the traditional diffusion-sorption model for bentonite and claystone. This model assumes that ion exchange binds cations to the solid, making them immobile, while diffusion occurs solely in a bulk water phase (which, incredibly, is assumed to fill the entire pore volume).
However, the idea that the exchanged ion is immobile does not agree with descriptions in the more general ion exchange literature, which instead acknowledge the process as an aspect of the Donnan effect.
Indeed, already in 1919, Donnan & Garner reported Na/K exchange equilibrium in a system consisting of two ferrocyanide solutions separated by a membrane impermeable to ferrocyanide, and it is fully clear that the particular distribution of cations in such systems is just as “thermodynamically necessary” as the distribution of chloride in the initial work on Congo Red and ferrocyanide.
Applied to clays, it is clear that cation exchange occurs even without postulating specific “sorption sites” or immobilization. On the contrary, ion exchange occurs in Donnan systems precisely because the ions are mobile.
In his book “Ion exchange”,10 Freidrich Helfferich describes ion exchange as diffusion, and distinguishes it from “chemical” processes
Occasionally, ion exchange has been referred to as a “chemical”
process, in contrast to adsorption as a “physical” process. This
distinction, though plausible at first glance, is
misleading. Usually, in ion exchange as a redistribution of ions by
diffusion, chemical factors are less significant than in adsorption
where the solute is held by the sorbent by forces which may not be
purely electrostatic.
Furthermore, in describing a general ion exchange system, he states the exact characteristics of a Donnan system, with the crucial point that the exchangeable ion is “free”, albeit subject to the constraint of electroneutrality
Ion exchangers owe their characteristic properties to a peculiar feature of their structure. They consist of a framework which is held together by chemical bonds or lattice energy. This framework carries a positive or negative electric surplus charge which is compensated by ions of opposite sign, the so-called counter ions. The counter ions are free to move within the framework and can be replaced by other ions of the same sign. The framework of a cation exchanger may be regarded as a macromolecular or crystalline polyanion, that of an anion exchanger as a polycation.
To give a very simple picture, the ion exchanger may be compared to a sponge with counter ions floating in the pores. When the sponge is immersed in a solution, the counter ions can leave the pores and float out. However, electroneutrality must be preserved, i.e., the electric surplus charge of the sponge must be compensated at any time by a stoichiometrically equivalent number of counter ions within the pores. Hence a counter ion can leave the sponge only when, simultaneously, another counter ion enters and takes over the task of contributing its share to the compensation of the framework charge.
With this “sponge” model at hand, he argues for that the reaction presented in eq. 2 above should be reformulated
[T]he model shows that ion exchange is essentially a statistical redistribution of counter ions between the pore liquid and the external solution, a process in which neither the framework nor the co-ions take part. Therefore Eqs. (1-1) [eq. 2 above] and (1-2) should be rewritten: \begin{equation} 2\overline{\mathrm{Na^+}} + \mathrm{Ca^{2+}} \leftrightarrow \overline{\mathrm{Ca^{2+}}} + 2\mathrm{Na^{+}} \end{equation} \begin{equation} 2\overline{\mathrm{Cl^-}} + \mathrm{SO_4^{2+}} \leftrightarrow \overline{\mathrm{SO_4^{2-}}} + 2\mathrm{Cl^{-}} \end{equation} Quantities with bars refer to the inside of the ion exchanger.
This “statistical redistribution” is of course nothing but the
establishment of Donnan equilibrium between the external solution and
the exchanger phase (as in the animation above). Naturally, Donnan
equilibrium — using either the “classical” or the “new” equations
— is at the heart of
manyanalyses of
ion exchange systems.
Unfortunately, this has not been the tradition in the compacted
bentonite research field, where a “diffuse layer” approach to cation
exchange has only been considered in more recent years, and then
usually as a supplement to already existing models and tools. We are
therefore in the rather uneasy situation that ion exchange in
bentonite nowadays often is explained in terms of both a Donnan
effect and as specific surface complexation.
Considering the robust evidence for significant ion mobility in interlayer pores, I strongly doubt surface complexation to be relevant for describing ion exchange in bentonite.11 Instead, I believe that not separating these processes obscures the analysis of species that actually do sorb in these systems. In any event, the exact effects of Donnan equilibrium — a mechanism dependent on nothing but that some charges diffuses through the semi-permeable component, while some others don’t — must first and foremost be worked out.
A demonstration of compacted bentonite as a Donnan system
To demonstrate how well the Donnan effect in compacted bentonite is captured by Donnan’s original description, we use the following relation, derived from eq. 1 (i.e we assume only the presence of a 1:1 salt, apart from the impermeable component)
Here \(z\) denotes the concentration of cations compensating impermeable charge. Eq. 3 quantifies anion exclusion, and is seen to depend only on the ratio \(c_\mathrm{Cl^-}^\mathrm{ext}/z\).
This equation is plotted in the diagram below, together with data of
chloride exclusion in sodium dominated bentonite
(Van Loon et
al., 2007) and in potassium ferrocyanide
(Donnan & Allmand,
1914)
I find this plot amazing. Although some points refer to bentonite at density 1900 \(\mathrm{kg/m^3}\) (corresponding to \(z \approx 5\) M), while others refer to a solution of approximately 25 mM \(\mathrm{K_4Fe(CN)_6}\) (\(z \approx 0.1\) M), the anion exclusion behavior is basically identical! Moreover, it fits the ideal “Donnan model” (eq. 3) quite well!
There is of course a lot more to be said about the detailed behavior of these systems, but I think a few things stand out:
It should be obvious that the basic mechanism for anion exclusion is the same in these two systems. This observed similarity thus invalidates the idea that anion exclusion in compacted bentonite is due to an intricate, ionic strength-dependent partitioning of a complex pore structure into parts which either are, or are not, accessible to chloride. In other words, the above plot is another demonstration that the concept of “accessible anion porosity” is nonsense.
The similarity between compacted bentonite and the simpler ferrocyanide system confirms Overbeek’s statement above, that Donnan’s “elementary” equations apply when the colloid concentration (i.e. density) is high enough.
The slope of the curve at small external concentrations directly reflects the amount of exchangeable cations that contributes to the Donnan effect. The similarity between model and experimental data thus confirms that the major part of the cations are mobile, i.e. not adsorbed by surface complexation. The similarity between the bentonite system and the ferrocyanide system also suggests that non-ideal corrections to the theory is better dealt with by means of e.g. activity coefficients, rather than by singling out a quite different mechanism (surface complexation) in one of the systems.
[1] The only equilibrium study of this kind I am aware of, that involves compacted, purified, homo-ionic clay, is Karnland et al. (2011). This study concerns Na/Ca exchange, and does not investigate the associated chloride equilibrium.
[2] I have assumed a K/Na selectivity coefficient of 2, and 95% salt exclusion.
[3] “Bentonite” is used in the following as an abbreviation for bentonite and claystone, or any clay system with significant cation exchange capacity.
[4] This particular publication states that I am one of the researchers using a “Donnan approach” to model “anion porosity”. Let me state for the record that I never have modeled “anion porosity”, or have any intentions to do so.
[6] In my head, a “soil suspension” and a “soil particle” are not very well defined entities. As I understand, Mattson investigated “Sharkey soil” and “Bentonite”. Sharkey soil is reported to have a cation exchange capacity of around 0.3 eq/kg, and the bentonite appear to be of “Wyoming” type. It is thus reasonably clear that Mattson’s “soil” particles are montmorillonite particles.
[7] Mattson and co-workers published a whole series of papers on “The laws of soil colloidal behavior” during the course of over 15 years, and appear to have caused both awe and confusion in the soil science community. I find it a bit amusing that there is a published paper (Kelley, 1943) which in turn reviews and comments on Mattson’s papers. Some statements in this paper include: “It seems to be generally agreed that some of [Mattsons papers] are difficult to understand.” and “The extensive use by [Mattson and co-workers] of terms either coined by them or used in new settings, the frequent contradictions of statement and inconsistencies in definition, and perhaps most important of all, the use by the authors of theoretical reasoning founded, not on experimentally determined data, but on calculations based on purely hypothetical premises, make it difficult to condense these papers into a form suitable for publication without doing injustice to the authors or sacrificing strict accuracy.”
[8] It may be worth noting that the only works referenced by Schofield — apart from a paper on dye adsorption — are Mattson, Procter and Donnan. Remarkably, Gouy is not referenced!
[10] In its introduction is found the following gem: “A spectacular evolution began in 1935 with the discovery by two English chemists, Adams and Holmes, that crushed phonograph records exhibit ion-exchange properties.” Who wouldn’t want to hear more of that story?!
[11] As a further argument for that the concept of immobile exchangeable ions in bentonite is flawed, one can take a look at the spread in reported values for the fraction of such ions. You can basically find any value between \(>99\%\) and \(\sim 0\%\) for the same type of systems. To me, this indicates overparameterization rather than physical significance.
Disclaimer: The following discussion applies fully to ions that only interact with bentonite by means of being part of an electric double layer. Here such ions are called “simple” ions. Species with more specific chemical interactions will be discussed in separate blog posts.
The “surface diffusion” model is not suitable for compacted bentonite
In the previous post on sorption1 we derived a correct “surface diffusion” model. The equation describing the concentration evolution in such a model is a real Fick’s second law, meaning that it only contains the actual diffusion coefficient (apart from the concentration itself)
\begin{equation} \frac{\partial c}{\partial t} = D_\mathrm{sd} \cdot\nabla^2 c \tag{1} \end{equation}
Note that \(c\) in this equation still denotes the concentration in the
presumed bulk water,2 while \(D_\mathrm{sd}\) relates
to the mobility, on the macroscopic scale, of a diffusing species in a
system consisting of both bulk water and surfaces.3
Conceptually, eq. 1 states that there is
no sorption in a surface diffusion model, in the sense that species do
not get immobilized. Still, the concept of sorption is frequently
used in the context of surface diffusion, giving rise to phrases such
as
“How Mobile Are Sorbed
Cations in Clays and Clay Rocks?”. The term “sorption” has
evidently shifted from referring to an immobilization process, to only
mean the uptake of species from a bulk water domain to some other
domain (where the species may or may not be mobile). In turn, the role
of the parameter \(K_d\) is completely shifted: in the traditional model
it quantifies retardation of the diffusive flux, while in a surface
diffusion model it quantifies enhancement of the flux (in a
certain sense).
A correct4 surface diffusion model resolves several of the
inconsistencies experienced when applying the traditional
diffusion-sorption model to cation diffusion in bentonite. In
particular, the parameter referred to as \(D_e\) may grow indefinitely
without violating physics (because it is no longer a real diffusion
coefficient), and the insensitivity of \(D_\mathrm{sd}\) to \(K_d\) may be
understood because \(D_\mathrm{sd}\) is the real diffusion
coefficient (it is not an “apparent” diffusivity, which is
expected to be influenced by a varying amount of immobilization).
Still, a surface diffusion model is not a very satisfying description of bentonite, because it assumes the entire pore volume to be bulk water. To me, it seems absurd to base a bentonite model on bulk water, as the most characteristic phenomenon in this material — swelling — relies on it not being in equilibrium with a bulk water solution (at the same pressure). It is also understood that the “surfaces” in a surface diffusion model correspond to montmorillonite interlayer spaces — here defined as the regions where the exchangeable ions reside5 — which are known to dominate the pore volume in any relevant system.
Indeed, assuming that diffusion occurs both in bulk water and on
surfaces, it is expected that \(D_\mathrm{sd}\) actually should
vary significantly with background concentration, because a diffusing
ion is then assumed to spend considerably different times in the two
domains, depending on the value of \(K_d\).6
Using the sodium diffusion data from Tachi and Yotsuji (2014) as an example, \(\rho\cdot K_d\) varies from \(\sim 70\) to \(\sim 1\), when the background concentration (NaCl) is varied from 0.01 M to 0.5 M (at constant dry density \(\rho=800\;\mathrm{kg/m^3}\)). Interpreting this in terms of a surface diffusion model, a tracer is supposed to spend about 1% of the time in the bulk water phase when the background concentration is 0.01 M, and about 41% of the time there when the background concentration is 0.5 M7. But the evaluated values of \(D_\mathrm{sd}\) (referred to as “\(D_a\)” in Tachi and Yotsuji (2014)) show a variation less than a factor 2 over the same background concentration range.
Insignificant dependence of \(D_\mathrm{sd}\) on background
concentration is found generally in the literature data, as seen here
(data sources:
1,
2,
3,
4,
5)
These plots show the deviation from the average of the macroscopically observed diffusion coefficients (\(D_\mathrm{macr.}\)). These diffusion coefficients are most often reported and interpreted as “\(D_a\)”, but it should be clear from the above discussion that they equally well can be interpreted as \(D_\mathrm{sd}\). The plots thus show the variation of \(D_\mathrm{sd}\), in test series where \(D_\mathrm{sd}\) (reported as “\(D_a\)”) has been evaluated as a function of background concentration.8 The variation is seen to be small in all cases, and the data show no systematic dependencies on e.g. type of ion or density (i.e., at this level of accuracy, the variation is to be regarded as scatter).
The fact that \(D_\mathrm{sd}\) basically is independent of background
concentration strongly suggests that diffusion only occurs in a single
domain, which by necessity must be interlayer pores. This conclusion
is also fully in line with the basic observation that interlayer pores
dominate in any relevant system.
where the concentration of the species under consideration, \(c^\mathrm{int}\), is indexed with an “int”, to remind us that it refers to the concentration in interlayer pores. The corresponding diffusion coefficient is labeled \(D_c\). Notice that \(c^\mathrm{int}\) and \(D_c\) refer to macroscopic, averaged quantities; consequently, \(D_c\) should be associated with the empirical quantity \(D_\mathrm{macr.}\) (i.e. what we interpreted as \(D_\mathrm{sd}\) in the previous section, and what many unfortunately interpret as \(D_a\)) — \(D_c\) is not the short scale diffusivity within an interlayer.
For species that only “interact” with the bentonite by means of being part of an electric double layer (“simple” ions), diffusion is the only process that alters concentration, and the continuity equation has the simplest possible form
Here \(n\) is the total amount of diffusing species per volume porous system, i.e. \(n = \phi c^\mathrm{int}\). Inserting the expression for the flux in the continuity equation we get
Eqs. 2 and 3
describe diffusion, at the Fickian level, in the homogeneous mixture
model for “simple” ions. They are identical in form to the Fickian
description in a conventional porous system; the only “exotic” aspect
of the present description is that it applies to interlayer
concentrations (\(c^\mathrm{int}\)), and more work is needed in order
to apply it to cases involving external solutions.
But for isolated systems, e.g. closed-cell diffusion
tests, eq. 3 can be applied directly. It is
also clear that it will reproduce the results of such tests, as the
concentration evolution is known to obey an equation of this form
(Fick’s second law).
The experimental data in this plot (from Sato et al. (1992)) represent the typical behavior of simple ions in compacted bentonite. The plot shows the resulting concentration profile in a Na-montmorillonite sample of density 600 \(\mathrm{kg/m^3}\), where an initial planar source of strontium, embedded in the middle of the sample, has diffused for 7 days. Also plotted are the identical results from fitting the three models to the data (the diffusion coefficient and the concentration at 0 mm were used as fitting parameters in all three models).
From the successful fitting of all the models it is clear that
bentonite diffusion data alone does not provide much information for
discriminating between concepts — any model which provides a
governing equation of the form of Fick’s second law will fit the
data. Instead, let us describe what a successful fit of each model
implies conceptually
The traditional diffusion-sorption model
The entire pore volume is filled with bulk water, in contradiction with the observation that bentonite is dominated by interlayer pores. In the bulk water strontium diffuse at an unphysically high rate. The evolution of the total ion concentration is retarded because most ions sorb onto surface regions (which have zero volume) where they become immobilized.
The “surface diffusion” model
The entire pore volume is filled with bulk water, in contradiction with the observation that bentonite is dominated by interlayer pores. In the bulk water strontium diffuse at a reasonable rate. Most of the strontium, however, is distributed in the surface regions (which have zero volume), where it also diffuse. The overall diffusivity is a complex function of the diffusivities in each separate domain (bulk and surface), and of how the ion distributes between these domains.
The homogeneous mixture model
The entire pore volume consists of interlayers, in line with the observation that bentonite is dominated by such pores. In the interlayers strontium diffuse at a reasonable rate.
From these descriptions it should be obvious that the homogeneous mixture model is the more reasonable one — it is both compatible with simple observations of the pore structure and mathematically considerably less complex as compared with the others.
The following table summarizes the mathematical complexity of the
models (\(D_p\), \(D_s\) and \(D_c\) denote single domain diffusivities,
\(\rho\) is dry density, and \(\phi\) porosity)
Note that the simplicity of the homogeneous mixture model is achieved
by disregarding any bulk water phase: only with bulk water absent is
it possible to describe experimental data as pure diffusion in a
single domain. This process — pure diffusion in a single domain — is
also suggested by the observed insensitivity of diffusivity to
background concentration.
These results imply that “sorption” is not a valid concept for simple cations in compacted bentonite, regardless of whether this is supposed to be an immobilization mechanism, or if it is supposed to be a mechanism for uptake of ions from a bulk water to a surface domain. For these types of ions, closed-cell tests measure real (not “apparent”) diffusion coefficients, which should be interpreted as interlayer pore diffusivities (\(D_c\)).
Footnotes
[1] Well, the subject was rather on “sorption” (with quotes), the point being that “sorbed” ions are not immobilized.
[2] Eq. 1 can be transformed to an equation
for the “total” concentration by multiplying both sides by
\(\left (\phi + \rho\cdot K_d\right)\).
[3] Unfortunately, I called this quantity
\(D_\mathrm{macr.}\) in the previous post. As I here compare several
different diffusion models, it is important to separate between
model parameters and empirical parameters, and the diffusion
coefficient in the “surface diffusion” model will henceforth be
called \(D_\mathrm{sd}\). \(D_\mathrm{macr.}\) is used to label the
empirically observed diffusion parameter. Since the “surface
diffusion” model can be successfully fitted to experimental
diffusion data, the value of the two parameters will, in the
end, be the same. This doesn’t mean that the distinction between the
parameters is unimportant. On the contrary, failing to separate
between \(D_\mathrm{macr.}\) and the model parameter \(D_\mathrm{a}\)
has led large parts of the bentonite research community to assume
\(D_\mathrm{a}\) is a measured quantity.
[5] There is a common alternative, implicit, and absurd definition of interlayer, based on the stack view, which I intend to discuss in a future blog post. Update (220906): This interlayer definition and stacks are discussed here.
[6] Note that, although \(D_\mathrm{sd}\) is not given simply
by a weighted sum of individual domain diffusivities in the surface
diffusion model, it is some crazy function of the ion
mobilities in the two domains.
[7] With this interpretation, the fraction of bulk water ions is given
by \(\frac{\phi}{\phi+\rho K_d}\).
[8] The plot may give the impression that such data is vast, but these are basically all studies found in the bentonite literature, where background concentration has been varied systematically. Several of these use “raw” bentonite (“MX-80”), which contains soluble minerals. Therefore, unless this complication is identified and dealt with (which it isn’t), the background concentration may not reflect the internal chemistry of the samples, i.e. the sample and the external solution may not be in full chemical equilibrium. Also, a majority of the studies concern through-diffusion, where filters are known to interfere at low ionic strength, and consequently increase the uncertainty of the evaluated parameters. The “optimal” tests for investigating the behavior of \(D_\mathrm{macr.}\) with varying background concentrations are closed-cell tests on purified montmorillonite. There are only two such tests reported (Kozaki et al. (2008) and Tachi and Yotsuji (2014)), and both are performed on quite low density samples.
In through-diffusion tests, diffusion is monitored from an external
source reservoir, through a clay sample, into an external target
reservoir. As the sample typically is sandwiched between two confining
filters, the full set-up can be abstracted as transport across three
conductive components, coupled in series (filter-clay-filter).
Solving this problem — which is not specifically related to diffusion
in clay, and applies equally well to e.g. electric currents or laminar
fluid flow — the steady-state flux can be written as (for details,
see appendix)
Here \(D_e\) denotes the effective diffusivity for the different
components1,
\(c_\mathrm{source}\) is the constant source reservoir concentration,
\(L_\mathrm{clay}\) is the length of the clay sample, and
\(L_\mathrm{filter}\) is the length of the filters (we assume that the
two filters have the same length).
Eq. 1 shows that in the limit \(\omega \ll 1\), the flux is expressed solely in terms of clay parameters2
It is thus clear that through-diffusion tests should be designed to
have \(\omega\) reasonably small; preferably, one should have
\(\omega \ll 1\), which allows for leaving the filters out of the
analysis.
While filter parameters in practice are limited to a quite small range for a given ion3, \(D_\mathrm{e}^\mathrm{clay}\) is known to grow indefinitely for many cations, as the background concentration tends to zero1. Consequently, for such ions, there always exists a background concentration limit, beyond which the filters completely control the resulting flux (i.e. where \(\omega \gg 1\)).
Even though the effect of filters in through-diffusion tests has been identified for a long time, there are numerous examples in the bentonite literature where filter influence is ignored, or not fully identified, leading to erroneous interpretations. For example, when describing through-diffusion tests with strontium in Boom Clay, Altmann et al. (2012) write
The resulting \(\alpha\) value of \(\sim 440\) corresponds to a \(K_d\) value similar to that measured on dispersed and intact Boom Clay. However, if this \(\alpha\) is used to estimate the corresponding \(D_e\) value via \(D_e = \alpha\cdot D_a\), the value obtained is \(\sim 45\) times higher than \(D_e(\mathrm{HTO})\), which is an unrealistically large difference. This is probably because the necessary conditions for calculating \(D_e\) by Fick’s law were not satisfied as indicated by the fact that the concentration profiles measured at the end of the through-diffusion experiment were unexpectedly ‘flat’, i.e. did not vary in a linear fashion between the surfaces in contact with the source and sink solutions. The reason for this behaviour is not yet known.
But a “flat” concentration profile is a key signature of filters
limiting the flux, as the (external) concentration difference across
the clay is (see appendix)
which approaches zero when \(\omega\) becomes large.
Consequently, the reported behavior strongly indicates that Boom Clay
has a very high transport capacity for strontium under the right
conditions (the test was performed with a sodium background
concentration of approximately 0.02 M), leading to the filters
limiting the flux. This, in turn, implies that the value for \(D_e\) in
the clay is underestimated, rather than being “unrealistically
large”.
What is demonstrated in this test — but not concluded — is that the principal diffusion mechanism in Boom Clay is the same as in compacted bentonite: ions assumed “sorbed” contribute to — and probably dominate — the diffusive flux. The traditional sorption-diffusion model is not valid for Boom Clay.
Glaus et al. (2007) clearly demonstrate filter influence on Na diffusion in Na-montmorillonite, performed over a large range of \(\mathrm{NaClO_4}\) background concentrations. The concentration profiles across the samples at the time of termination look like this4
The profiles become increasingly “flat” with decreasing background
concentration, demonstrating an increasing transport capacity of the
clay (demonstrating this transport capacity was the main purpose of
the study). The tests in
Glaus et al. (2007)
are analyzed assuming a filter-clay-filter configuration, with
identical diffusivities for the two filters (for a given test). The
clay component is described using the traditional sorption-diffusion
model5. From the reported
fitted model parameters, we can calculate the corresponding relative
filter resistances, using eq. 2. The result is as follows (all these
samples have \(L_\mathrm{clay}=5.4\) mm and \(L_\mathrm{filter}=1.55\)
mm.)
\(C_\mathrm{bkg}\)
(\(\mathrm{mol/m^3}\))
10
100
500
700
1000
Reported
\(D_\mathrm{e}^\mathrm{clay}\)
(\(10^{-10}\;\mathrm{m^2/s}\))
14
3.7
0.86
0.53
0.38
\(D_\mathrm{e}^\mathrm{filter}\)
(\(10^{-10}\;\mathrm{m^2/s}\))
0.86
1.0
0.86
0.86
0.86
Calculated
\(\omega\)
(-)
9.3
2.1
0.6
0.4
0.3
Indeed, \(\omega \gg 1\) for the test performed at 10 mM
\(\mathrm{NaClO_4}\), and filters fully control the flux. Filter
influence is also significant in the test at 100 mM (\(\omega =
2.1\)), while the effect is less important in the tests at higher
background concentration. These results fully reflect the appearance of
the concentration profiles above.
The filter influence is also clearly seen in the behavior of the
outfluxes at the different background concentrations (dotted graphs)
For the tests at high background concentration (i.e. small \(\omega\)),
steady-state6 is
reached in about 8 – 10 days. In the 10 mM-test, on the other hand,
the system is far from steady-state even after 45 days7 — the outflux is still increasing, even though
the source concentration (dash-dotted graphs) has dropped
significantly. A prolonged transient state is thus another key
signature of filters limiting the flux.
This prolonged transient appears because the clay has to be “filled up” with ions before a steady-state can be established. It is important not to confuse this effect with that of retardation due to increased “sorption”: here, it is the filters that cannot “fill up” the clay fast enough, while the diffusive transport capacity of the clay actually increases with increasing “sorption”. Note that this increased transport capacity is not due to increased diffusivity, but exactly because the clay accommodates an increasing amount of tracers as the background concentration decreases.
For the most part, Glaus et al. (2007) treat the filter influence adequately, allowing them to draw correct conclusions regarding diffusion in compacted bentonite. Going into detail, however, I think there is some inconsistency in the parameters, demonstrating the inherent difficulties with handling cation through-diffusion at low ionic strength. \(K_d\) has, as far as I see, been used as a free fitting parameter in the modeling of the tests.8 But for the specific case of sodium tracers diffusing in pure sodium montmorillonite, this parameter is constrained by the simple relation (which also is derived in the article)
where \(C_{bkg}\) denotes the background concentration, and CEC is the
cation exchange capacity. The reported \(K_d\) values, thus corresponds
to these CEC values
\(C_\mathrm{bkg}\)
(\(\mathrm{mol/m^3}\))
10
100
500
700
1000
Reported
\(K_d\)
(\(10^{-3}\) \(\mathrm{m^3/kg}\))
46
7.3
1.8
1.2
0.74
Calculated
CEC
(eq/kg)
0.46
0.73
0.90
0.84
0.74
As the documented CEC for the used material (purified “Milos”
montmorillonite)
is
\(\sim 0.88\) eq/kg, this evaluation indicates that the fitted \(K_d\) is
significantly underestimated for the test performed at 10 mM.
The reason for this underestimation can be further investigated by using the end values of the recorded clay concentration profile, and assuming the CEC value (i.e. assuming \(K_d\), using eq. 3). From the definition of \(K_d\) we can thereby calculate \(c_\mathrm{in}\) and \(c_\mathrm{out}\).
\(C_\mathrm{bkg}\)
(\(\mathrm{mol/m^3}\))
10
100
500
700
1000
Reported
\(s_{in}\)
(\(10^{-12}\) mol/kg)
88.5
38.5
12.2
9.3
7.8
\(s_{out}\)
(\(10^{-12}\) mol/kg)
76.3
17.4
1.4
1.1
\(\sim 0\)
\(c_{source}\)
(\(10^{-9}\) \(\mathrm{mol/m^3}\))
3.1
10.5
8.1
7.7
9.4
Assumed
\(K_d\)
(\(10^{-3}\) \(\mathrm{m^3/kg}\))
88
8.8
1.76
1.26
0.88
Calculated
\(c_{in}\)
(\(10^{-9}\) \(\mathrm{mol/m^3}\))
1.0
4.4
6.9
7.4
8.9
\(c_{out}\)
(\(10^{-9}\) \(\mathrm{mol/m^3}\))
0.9
2.0
0.8
0.9
\(\sim 0\)
\(\omega\)
(-)
21.36
3.37
0.31
0.19
\(\sim 0\)
This calculation gives a concentration drop across the inlet filter
(\(c_\mathrm{source} – c_\mathrm{in}\)) that is considerably larger than
half the value of the concentration in the source reservoir
(\(c_\mathrm{source}\)), for the tests made at 10 mM and 100 mM. Such a
behavior is impossible if the diffusivities of the two filters are
identical! This reevaluation thus suggests that it is not strictly
valid to assume identical filter diffusivities when evaluating these
kinds of tests. Of course, if the tests are performed under
conditions with small \(\omega\), this assumption will make little
difference, because the filter influence is anyway small. But under
conditions with \(\omega \gg 1\), the exact values of both filter
diffusivities will significantly influence the analysis. The
concentration profiles across the filters in the 10 mM case can be
illustrated like this
The main achievement in Glaus
et al. (2007) is that they, despite filter transport complications,
manage to verify that the effective diffusivity in the clay, both for
sodium and strontium tracers, scale with sodium background
concentration as
where \(Z\) is the valency of the tracer (i.e., \(Z = 1\) for sodium, and \(Z = 2\) for strontium). Not only is this relation crucial for understanding bentonite diffusion at a deeper level, it also allows for assessing filter influence on evaluated diffusion parameters in general. Eq. 4 implies a dramatic effect of the background concentration on the relative filter resistance for strontium (note from eq. 2 that also \(\omega\) will scale as \(C_\mathrm{bkg}^{-Z}\)): lowering the background concentration e.g. from 0.5 M to 0.1 M, increases \(\omega\) by a factor of 25; lowering it from 0.5 M to 0.01 M gives a factor of 2500! (I don’t think it is a coincidence that the strontium tests in Glaus et al. (2007) are restricted to \(C_\mathrm{bkg}\ge 0.5\;\mathrm{M}\).)
Molera and Eriksen (2002) report diffusion parameters evaluated for strontium in “MX-80” bentonite of various densities and in the background concentration (\(\mathrm{NaClO_4}\)) range 0.5 M – 0.01 M. The tests were evaluated using the traditional sorption-diffusion model for the clay, and by taking the filters into account. The filter diffusivities were, however, assumed identical in the two filters, and kept constant (for a given ion) in all models. From the reported fitted parameters, we can evaluate \(\omega\), using eq. 2 (they used “\(D_\mathrm{a}\)” as fitting parameter rather than \(D_\mathrm{e}^\mathrm{clay}\), but these are related via \(D_\mathrm{e}^\mathrm{clay} = D_\mathrm{a}\left(\phi + \rho K_d\right)\))
Reported
Calculated
Density
\(C_\mathrm{bkg}\)
\(D_\mathrm{a}\)
\(K_d\)
\(D_\mathrm{e}^\mathrm{clay}\)
\(\omega\)
(\(\mathrm{kg/m^3}\))
(\(\mathrm{mol/m^3}\))
(\(10^{-10}\;\mathrm{m^2/s}\))
(\(10^{-3}\;\mathrm{m^3/kg}\))
(\(10^{-10}\;\mathrm{m^2/s}\))
(-)
400
100
0.43
110
19.3
6.8
800
100
0.35
150
42.2
14.8
800
500
0.40
15
5.1
1.8
1200
10
0.21
500
126
44.2
1200
100
0.18
130
28.2
9.9
1200
500
0.25
13
4.0
1.4
1600
10
0.14
1200
269
94.2
1600
100
0.10
90
14.4
5.1
1600
500
0.20
15
4.9
1.7
1800
100
0.09
80
13.0
4.6
1800
500
0.12
15
3.3
1.1
In this evaluation is used
\(D_\mathrm{e}^\mathrm{filter} = 0.925 \cdot 10^{-10}\;\mathrm{m^2/s}\),
\(L_\mathrm{filter} = 0.81\) mm, and \(L_\mathrm{clay} = 5.0\) mm for all
tests.
Filter transport dominates (\(\omega \gg 1\)) in all but the tests performed at 500 mM (and even in these tests, there is significant filter influence). It can therefore be questioned whether the parameters have been adequately evaluated. That the fitted parameter values (\(K_d\) and/or \(D_\mathrm{a}\)) are not adequate is seen when plotting \(D_\mathrm{e}^\mathrm{clay}\) against background concentration (the “expected dependency” line assumes the \(D_\mathrm{e}^\mathrm{clay}\) value of the 1200 \(\mathrm{kg/m^3}\) sample at 500 mM background concentration).
The \(D_\mathrm{e}^\mathrm{clay}\) values do not obey Glaus’ relation, which they are expected to do, as “MX-80” is a sodium dominated clay. Note that the above plot suggests that \(D_\mathrm{e}^\mathrm{clay}\) in Molera and Eriksen (2002) at background 0.01 M may be underestimated by roughly two orders of magnitude! Nevertheless, the actual clay diffusivity estimated in this study (unfortunately interpreted as “apparent” diffusivity) compares relatively well with other measurements, e.g. Kim et al. (1993), indicating that the underestimation of \(D_\mathrm{e}^\mathrm{clay}\) is rooted in a similar underestimation of \(K_d\).
Results like those of Glaus et al. (2007) and Molera and Eriksen (2002) show that cation through-diffusion tests at low background concentration should be avoided if possible: Both studies explicitly take into account filters when evaluating model parameters, yet the evaluations can be demonstrated to be inconsistent in the low background concentration limit. Although experimental design — as well as corresponding modeling — can be of various quality, the low concentration limit is fundamental: no matter how rigorous the analysis, the results will still be uncertain, simply because the experiment itself conveys less and less information on transport parameters in the clay.
Thus, unless the explicit purpose is to explore the low background concentration limit, it is better to stay away from it, thereby reducing the risk of drawing incorrect conclusions. An example of using data influenced by filter resistance to draw far-reaching conclusions regarding bentonite structure is found in Tinnacher et al. (2016).
This study uses the result from a single through-diffusion test in pure Na-montmorillonite (prepared from SWy-2) at 800 \(\mathrm{kg/m^3}\)9 to review “single porosity models”, and to argue for that this system is dominated by bulk water (\(>70\%\)) — a rather bizarre conclusion, in my opinion.
The test was done with a background electrolyte of 0.1 M NaCl, by adding a small amount of \(\mathrm{CaBr_2}\) (1 mM) to the source reservoir, and monitoring the accumulation of calcium and bromide in the target reservoir (which was kept virtually tracer free by frequent replacement). The recorded normalized outflux of calcium looks like this10
As this test concerns diffusion of a di-valent cation at relatively low ionic strength, there are strong reasons to suspect that filter resistance influences the flux evolution. If I understand correctly, this test was actually performed using the exact same equipment as used in the study by Molera and Eriksen (2002), where we evaluated a value \(\omega = 14.8\) for strontium at the same conditions, albeit in a different clay material (see above).
But using the reported model parameters in Tinnacher et al. (2016) gives \(\omega = 0.77\) (\(D_\mathrm{e}^\mathrm{clay} = 2.06\cdot 10^{-10}\; \mathrm{m^2/s}\), \(D_\mathrm{e}^\mathrm{filter} = 0.85\cdot 10^{-10}\; \mathrm{m^2/s}\), \(L_\mathrm{filter} = 0.79\) mm, and \(L_\mathrm{clay} = 5\) mm). This result — indicating only moderate filter influence — is a bit surprising, given the results from Molera and Eriksen (2002), and given that calcium appears to diffuse faster than strontium in Na-montmorillonite.
However, these model parameters are not consistent with the recorded
steady-state flux. The normalized flux (\(j/c_\mathrm{source}\)) can
be calculated from eq. 1
which is significantly smaller than the observed flux of
\(3.5 \cdot 10^{-8} \;\mathrm{m/s}\). In order to match the observed
flux instead requires
\(D_\mathrm{e}^\mathrm{clay} = 5.0\cdot10^{-10}\; \mathrm{m^2/s}\),
indicating significant filter influence after all (\(\omega = 1.86\)).
Of course, the calculated flux could match the observed flux by
instead altering the filter diffusivity (or by altering both the
filter and clay diffusivities). But matching the fluxes by only
altering the filter diffusivity requires
\(D_\mathrm{e}^\mathrm{filter} = 3.67\cdot 10^{-10}\; \mathrm{m^2/s}\),
which is unrealistically large (it corresponds to a geometric factor
of unity and porosity 0.46).
This analysis shows that the evaluated value for \(D_\mathrm{e}^\mathrm{clay}\) for calcium in Tinnacher et al. (2016) is conditioned on the adopted value for filter diffusivity, and that the experiment most probably is significantly influenced by filter limitations. It is consequently not suited for reviewing “single porosity models”.11
Footnotes
[1] Note that for bentonite, \(D_\mathrm{e}^\mathrm{clay}\) is not a real diffusion coefficient! But, since it is the parameter that quantifies the steady-state flux given the external concentration difference (\(c_\mathrm{in} – c_\mathrm{out}\)), it is precisely what is required in this analysis.
[2] Except for \(c_\mathrm{source}\), of course; without a source
concentration there wouldn’t be much flux.
[3] Typically, \(L_\mathrm{filter} \sim 1\) mm and \(D_\mathrm{e}^\mathrm{filter} \sim 0.1\cdot D_0\), where \(D_0\) is the corresponding diffusivity in pure bulk water.
[4] The data underlying these plots are found in the supporting information to Glaus et al. (2007). There it is, however, presented as “normalized” concentrations, without a full description of how this normalization has been performed. I have used the concentration values as plotted, but scaled them spatially to the proper sample length (5.4 mm).
[5] In contrast to basically any other diffusion study, the traditional model is (in a sense) concluded invalid in Glaus et al. (2007). For this reason, the quantity usually labeled \(D_\mathrm{e}\) is in this paper labeled \(^cD\), where “c” is short for “conditional”. Here, we continue to label this quantity \(D_\mathrm{e}^\mathrm{clay}\), in order to relate it to other studies.
[6] These tests were performed with a changing
source reservoir concentration (also plotted), and the system is
never strictly in steady-state, as reflected in a weak decay of the
flux at long times. Still, there is a distinct difference between
this “quasi”-steady-state and the initial transient state, and the
presented theoretical analysis is still useful to apply.
[7] The supporting information unfortunately leaves out the data
between days 45 and 100.
[9] Tinnacher et al. (2016) states the density as both 800 \(\mathrm{kg/m^3}\) and 790 \(\mathrm{kg/m^3}\). I have used the former value.
[10] Oddly, the “flux” data is presented in Tinnacher et al. (2016) without correcting for a certain amount of “dead” volume that is not being exchanged during the target concentration measurements. Consequently, what is called “flux” in the article is strictly not the real flux, and all model curves look like a hedgehog’s back. In the plot presented here, this correction has been performed, and it does not exactly resemble the published plot. In practice, these differences are not important for the point I’m trying to make: the steady-state flux is still the same.
[11] I mean that diffusion
studies in general are not very useful on their own for drawing
conclusions on e.g. the presence of bulk water in bentonite. But
that’s a separate discussion.
Appendix: Derivation of eqs. 1 and 2
We assume that the steady-state flux in any of the conductive units is
linearly dependent on the concentration difference applied across it
(\(\Delta c\))
\begin{equation}
j = -\frac{1}{R}\Delta c
\end{equation}
where \(R\) is the transfer resistance.
With constant source and target concentrations, the steady-state flux
in the system under consideration (filter-clay-filter) can be
expressed using any of the involved units (the flux is the same
everywhere)
Disclaimer: The following discussion applies fully to ions that only interact with bentonite by means of being part of an electric double layer. Species with more specific chemical interactions will be discussed in separate blog posts.
The entire pore volume contains bulk water, where ions diffuse according to \begin{equation} j = -D_e \nabla c \tag{1} \end{equation} where \(c\) is the ion concentration and \(D_e = \phi D_p\), with \(\phi\) being the porosity, and \(D_p\) a so-called pore diffusivity (containing information on the ion mobility at the macroscopic scale of the porous system).
Ions in the porewater “sorb” on solid surfaces and become immobilized. The amount of “sorbed” ions per unit solid mass, \(s\), is related to \(c\) via a distribution coefficient \(K_d\) \begin{equation} s = K_d\cdot c \tag{2} \end{equation}
With these assumptions, the total amount of ions in the bentonite
(per volume porous medium) is
\begin{equation}
n = \phi\cdot c + \rho\cdot s = \left (\phi+\rho K_d \right) c
\tag{3}
\end{equation}
where \(\rho\) is the (dry) density of the clay. Applying the continuity
equation, \(\partial n/\partial t = -\nabla\cdot j\), gives
The model defined by eqs. 5
and 6 — which we will refer to as the
traditional model — has a distinct physical interpretation: ions
diffuse relatively fast in a bulk water phase, while being retarded
due to “sorption” onto the solid
surfaces. Although eq. 5 has the form of
Fick’s second law, it is clear that \(D_a\) is not a real
diffusion coefficient, but is influenced both by diffusion (\(D_e\)) and
“sorption” (\(K_d\)).
The immobilization assumption is not valid
The traditional model has a major problem: it is not valid for compacted bentonite. There is — and was — strong evidence that the immobilization assumption does not hold for all types of “sorbing” ions. E.g Jenny and Overstreet (1939) reported transport of \(\mathrm{Fe^{2+}}\), \(\mathrm{H^+}\), \(\mathrm{K^+}\), \(\mathrm{Na^+}\), and \(\mathrm{Ca^{2+}}\) as samples of H- K- Na- and Ca-bentonite was brought in contact with samples of Fe/H-bentonite (without gel contact there was no transport), and van Schaik et al. (1966) demonstrated steady-state fluxes of \(\mathrm{Na^+}\) through Na-montmorillonite, corresponding to pore diffusivites significantly larger than the \(\mathrm{Na^+}\) diffusivity in pure water. Moreover, ion mobility is an essential component of the most well-established theoretical concept used for describing montmorillonite — the electric double layer. The ionic part of the electric double layer is even referred to as the diffuse layer, for crying out loud!2
So it should come as no surprise that the pioneers in radwaste barrier research quickly came to the conclusion that the description underlying eq. 6 does not hold. Severalresearchgroups reported values of \(K_d\) and \(D_a\) that gives unreasonably large values of \(D_e\) when evaluated using eq. 6 (even larger than the corresponding diffusivities in pure water, in several cases).
What is more surprising is that these conclusions did not lead to a complete reevaluation of the underlying assumptions, which is the preferred procedure for a model producing nonsense. Instead, the invalidity of the traditional model was only considered a problem for the specific ions for which it yield “unrealistic” parameter values (in particular, strontium and cesium), and an adopted “remedy” was the so-called surface diffusion model. The surface diffusion model keeps the description expressed by eqs. 1, 2, 5, and 6, while replacing the relation \(D_e = \phi\cdot D_p\) with the following monster
where \(D_s\) is a so-called surface diffusion coefficient.
This extension of the traditional model brings about several problems,
the most glaring one being that eq. 7 is wrong. Therefore, to be able
to continue the discussion, we first have to derive a correct
“surface diffusion”3
model.
A correct “surface diffusion” model
Eq. 7 is not valid because it relies on the incorrect assumption that diffusive fluxes from different domains are additive. To derive the correct equations, we go back to the only conceptual modification made in the surface diffusion model: that the “sorbed” ions are no longer assumed immobile. Thus, the basic assumptions of the surface diffusion model are:
The entire pore volume contains bulk water, where ions diffuse.
Ions in the porewater “sorb” on the solid surfaces.
“Sorbed” ions also diffuse, along the surfaces.
There are consequently two domains (bulk water and surfaces) where
diffusion is assumed to take place, and a correct equation for the
macroscopic flux is in this case (for details, see
here)
\begin{equation}
j = -\phi D_\mathrm{macr.} \frac{\bar{c}}{c}\nabla c \tag{8}
\end{equation}
where \(D_\mathrm{macr.}\) is the diffusion coefficient on the macroscopic scale, which conveys information on the mobility in both of the involved domains, as well as their geometrical configuration. \(\bar{c}\) is the average concentration of all mobile entities; here, this include all ions — those in the porewater as well as those on the surfaces. The bulk water concentration is still labelled \(c\). Using eq. 3, we can write
\begin{equation}
\phi\bar{c} = n = \left (\phi + \rho K_d \right) c \tag{9}
\end{equation}
Further, when using eq. 10 in the continuity equation, the factor
\(\left (\phi + \rho K_d \right)\) cancels, giving
\begin{equation}
\frac{\partial c}{\partial t} = D_\mathrm{macr.} \nabla^2 c \tag{12}
\end{equation}
A complete conceptual change
Eqs. 11 and 12 are the main result of a correctly derived “surface
diffusion” model. Comparing with eqs. 5 and 6, we see that, although
the equations are deceptively similar, a “surface diffusion” model
brings about a complete conceptual change: with the immobilization
assumption released, it is actually the real diffusion
coefficient that appear in Fick’s second law (eq. 12). In fact,
the surface diffusion model does not contain any apparent
diffusivity!
Similarly, \(D_e\) is not a real diffusion coefficient in the surface diffusion model, as clearly seen by eq. 11. Rather, \(D_e\) is the product of the diffusion coefficient and a concentration factor, stemming from the fact that the general flux equation reads \(j = -\frac{D}{RT} \bar{c}\cdot \nabla \mu\), where \(\mu\) is the chemical potential of the diffusing ion.
In the same vein, \(K_d\) does not quantify “sorption” — at least if “sorption” is supposed to refer to a process that retards the diffusive flux. On the contrary, \(K_d\) acts as a kind of amplifier of the flux (eq. 10). This implication was already pointed out by Jahnke and Radke (1985):
Thus, \(K_d\) values are of extreme importance during the steady release period. Counter to what may have been anticipated, larger \(K_d\) values lead to higher release rates.
The conceptual changes of a “surface diffusion” model make a lot of
sense when interpreting diffusion data:
\(D_e\) is expected to vary with \(K_d\) and can basically grow without limit, without becoming unphysical.
“\(D_a\)” for a whole bunch of cations is universally found to beinsensitive tochanges in \(K_d\), even though \(K_d\) may vary orders of magnitudes (by altering the background concentration). This is the expected behavior of a real diffusion coefficient, and it makes all sense to instead interpret this parameter as \(D_\mathrm{macr.}\), using eq. 12.
When assuming all ions in the system to be mobile, we expect the governing equation (eq. 12) to be a real Fick’s second law, i.e. that it contains nothing but the actual diffusion coefficient.
Despite this sense-making, I have never seen these points being spelled out in the bentonite literature. Rather, it appears as the implication of releasing the immobilization assumption has not been fully comprehended. To this day, the traditional model is used as the basis for interpretation in basically all publications, to the extent that “\(D_a\)”, “\(D_e\)”, and “\(K_d\)” usually are considered to be experimental quantities. Similarly, most modern model development is focused on extending the traditional model, using flawed concepts regarding diffusivity in multi-porous systems.
I also find it symptomatic that “surface diffusion” not uncommonly is treated as being an optional mechanism, or even being completely discarded, while the dominant contribution of “sorbed” ions to the diffusive flux seems to have to be rediscovered every25thyear, or so.
Note that the conclusions made here are not based on any model that I favor, but are the consequence of a correct conceptual treatment of what is stated in the contemporary bentonite literature. Although a “surface diffusion” model (correctly treated!) make much more sense as compared with the traditional model in interpreting cation tracer diffusion, it still involves an assumption that I don’t understand how anybody can endorse: it assumes the entire pore volume to consist of bulk water. I will discuss this point in a separate blog post. Update (210225): Letting go of the bulk water is discussed here.
Footnotes
[1] In the following I will write “bentonite”, but I mean any type of clay system with significant ion exchange properties.
[2] I guess a reason for not acknowledging interlayer
diffusivity could have been the incorrect notions that interlayer
water is crystalline, and that
diffuse layers only “form” at higher water content.
[3] Since the established surface diffusion
model is that expressed by eq. 7, I use quotation marks when
speaking about models that don’t comply with this equation.
In the beginning there was the Poisson-Boltzmann equation. Solving it for the case of a salt solution in contact with a negatively charged plane surface (a.k.a. the Gouy-Chapman model) gives the concentration of cations and anions in the solution as a function of the distance to the surface, like this1
Note:
The suppression of the anion concentration near the surface is often referred to as negative adsorption or anion exclusion. The total amount of excluded anions per unit surface area (indicated in green), usually labeled \(\Gamma^-\), is obtained by integrating the Poisson-Boltzmann equation.
There are, nevertheless, anions everywhere! This model will give zero anion concentration only for an infinitely negative electrostatic potential (or if \(c_0 = 0\), of course).
A clever way to utilize negative adsorption is for estimating the amount of smectite surface area in a soil sample, first suggested by Schofield (1947). This is done by comparing measured values of negative adsorption with the appropriate expression evaluated from the Gouy-Chapman model. When doing the necessary math2 for such an analysis you naturally end up with expressions like
where \(c_0\) is the external anion concentration (i.e. far from the surface), and \(\kappa^{-1}\) is the Debye length. This equation, having the dimension of length, can be interpreted as the width, \(d_{ex}\), of a region devoid of anions, which gives the same amount of negative adsorption as the full exclusion region, as illustrated here (yellow)
However, note:
This is just an equivalent, fictitious region.
Anions are still everywhere!
Due to its convenience in the analysis, the notion of an equivalent
region devoid of anions — often referred to in terms of “volume of
exclusion” — became rather popular. At the same time, authors
stopped emphasizing that this is a fictitious region. A clear example
of such a transition is
Edwards and Quirk
(1962) who states that \(\Gamma^-/c_0\) “can be regarded as the
surface depth from which chloride ions are excluded”, while in
Edwards et al. (1965) the
same quantity (multiplied by area) is referred to as “the volume from
which chloride is excluded”. The latter statement is, strictly
speaking, wrong: the actual volume from which anions are excluded is
the entire region where the concentration deviates from \(c_0\), and the
exclusion is only partial — there are anions everywhere!
Compacted bentonite
But the idea of an actual region devoid of anions seems to have stuck, and I believe that this influenced the interpretation of diffusion in compacted bentonite3 in terms of “effective porosity” or “anion accessible-porosity”. Concepts which, in turn, have motivated the idea that bentonite contains bulk water (“free water”, “pore water”).
The first example of this usage in studies of compacted bentonite, that I know of, is in Muurinen et al. (1988) reporting chloride through-diffusion in bentonite with various densities and background concentrations.
The tracer concentration of the porewater clearly depends on the compaction of bentonite and on the salt concentration of the circulating water. The effective porosity can even be less than one percent when the salt concentration is low and compaction high. Also, the diffusivities strongly depend on the density of bentonite and on the salt concentration.
The low tracer concentration in bentonite in the diffusion tests […] are indicative of ion-exclusion [5]. Ion-exclusion probably decreases the effective size of the pores, which changes the geometric factor, of bentonite and thus the apparent diffusivity. In addition to the geometric factor, the effective diffusivity takes into account the effective pore volume; thus, the dependence is even stronger.
“Effective porosity” has not been defined earlier in the article, so it is difficult to know precisely what the authors mean by the term. But it is relatively clear4 from the second paragraph that they explain the measured fluxes as being a result of a physical variation of the pore volume accessible to anions, rather than as a variation of the tracer concentration in a homogeneous system. This is also supported by their writing in the conclusions section: “The decreased pore size and porosity caused by ion-exclusion could at least qualitatively explain the dependence.”
However, the reference they provide (“[5]”) is Soudek et al. (1984), who calculate anion exclusion by means of — the Poisson-Boltzmann equation! (Which predicts anions everywhere.) In fact, Soudek et al. (1984) calculate what they term “Donnan exclusion” in a homogeneous model of “parallel, equally-spaced platelets”. Thus, the reference supplied by Muurinen et al. (1988) is in direct contradiction with their interpretation that the pore size and porosity is decreasing with the salt concentration.
Soudek et al. (1984) even provide an example of how the average chloride concentration between the platelets depends on the separation distance, when in equilibrium with an external solution of 10 mM, and write
Note the extremely strong rejection of the co-ion. At 50 w% clay
(\(\sim 25\)Å plate separation) almost 90% of the anions are
rejected.
which is completely in line with the observation of Muurinen et al. (1988) that “The effective porosity can even be less than one percent when the salt concentration is low and compaction high”, if only “effective porosity” is replaced by “concentration between the plates”.
It makes me a bit tired to discover that the record could have been set straight over 30 years ago regarding which pores anions can access. Instead the bentonite research community, for the most part, doubled down on the idea that anions only have accesses to parts of the pore volume, or that compacted bentonite contains a significant amount of bulk water.
An explicit description of interpreting “chloride through diffusion
porosity” as a specific, limited part of the pore volume is given by
Bradbury and
Baeyens (2003)
In the interlayer spaces and regions where the individual montmorillonite stacks are in close proximity, double layer overlap will occur and anion exclusion effects will take place. Exclusion will probably be so large that it is highly unlikely that anions can move through these regions (Bolt and de Haan, 1982). However, Cl anions do move relatively readily through compacted bentonite since diffusion rates have been measured in ‘‘through-diffusion’’ tests […]
If the Cl anions cannot move through the interlayer and overlapping double layer regions because of anion exclusion effects, then it is reasonable to propose that the ‘‘free water’’ must provide the diffusion pathways (Fig. 1). Therefore, the hypothesis is that the pore volume associated with the transport of chloride (and other anions) is the ‘‘free water’’ volume, and that this is the porewater in a compacted bentonite.
Here they refer to Bolt and De Haan, (1982)5 when arguing for that anions do not have access to interlayers. But the analysis in this reference is based on nothing but — the Poisson-Boltzmann equation! (which predicts anions everywhere)
Another thing to note is the notion of “overlapping” diffuse layers. Studies of negative adsorption to quantify surface area typically look at soil suspensions, with a solid part of a few percent. In such systems it is justified to perform the analysis on a single diffuse layer because the distance between separate montmorillonite particles is large enough. But at higher density there is not enough space between separate clay particles for the ion concentrations to ever reach the “external” value (\(c_0\)) — the diffuse layers “overlap”.6
It has been shown that effects of “overlapping” diffuse layers on the resulting negative adsorption is significant already at a a solid content of 6%. When carrying over the anion exclusion analysis to compacted bentonite — with solid content typically above 70%! — it therefore becomes near impossible to believe that the system should contain regions unaffected by the montmorillonite (“free water”). Yet, the argumentation above, apart from being flawed in the way it refers to the Poisson-Boltzmann equation, relies critically on the existence of such regions.
Despite the improbability that montmorillonite particles in compacted bentonite can be spaced so far apart as to allow for bulk water within the system, the idea of anions only having access to “free” water was nevertheless further pursued by Van Loon et al. (2007). They provide a picture similar to this
The idea here (and elsewhere) is that bentonite consists of “stacks” of individual montmorillonite particles (TOT-layers) interlaced with interlayer water.7 The space between “stacks” is assumed large enough for diffuse layers to fully develop, and to merge into a bulk solution (“free water”), whose volume depends on the ionic strength, reminiscent of the excluded volume in eq 1.8 Anions are postulated to only have access to this “free” water.
But as references for anion exclusion is once again simply given studies based on the Poisson-Boltzmann equation (in particular, Bolt and De Haan, (1982)). But these — as I hope has been made clear by now — predict anions everywhere, and consequently do not support the suggested model. In this case, the mismatch between model and supporting references stands out, as the term “effective porosity” is used interchangeably with the term “Cl-accessible porosity”; if Gouy-Chapman theory in a convoluted way can be used to define an “effective” porosity (having no other meaning than a fictitious, equivalent volume), there is no possibility whatsoever to use it to support the idea of anions having access to only parts of the pore space. Ironically, “anion-accessible porosity” seems to be the most popular termnowadays for describingeffects of anionexclusion in compactedbentonite.
The strongest confirmation that the modern-day concept of anion-accessible porosity is simply a misuse of the exclusion-volume concept is given in Tournassat and Appelo (2011). They provide a quite extensive background for the type of anion exclusion they consider, and it is based on the excluded-volume concept discussed above. They even explicitly calculate the excluded-volume (named “total chloride exclusion distance”) only to directly discard it as not suitable
However, this binary representation (absence or presence of
chloride, Fig. 3) is not very representative of the system since the
EDL is not completely devoid of anions.
Yet, after making this statement that anions are everywhere (in the diffuse layer) they anyway define anion accessible porosity as an effective, fictitious volume!9
Due to the very narrow space, the double layers in the interlayers
overlap and the electric potential in the truncated layer becomes
large leading to a complete exclusion of anions from the interlayer
(Bolt and de Haan, 1982; Pusch et al., 1990; Olin, 1994; Wersin et
al., 2004). The interlayer water thus contains exclusively cations
that compensate the permanent charges located in the octahedral
layer of the clay.
When the dry density is above \(1.8 \;\mathrm{kg/dm^3}\), almost all the porosity resides in the interlayers of Na-montmorillonite. Since anions are excluded from the interlayers, the anion-accessible porosity becomes zero, and anion-diffusion is minimal (Bourg et al., 2003)
But in Bourg et al. (2003) is explicitly stated that anion exclusion from interlayers is only “partial”!
To sum up…
The idea that anions have access only to parts of the pore volume is
widespread in today’s compacted bentonite research community. In this
blog post I have shown that this idea emerges from misusing the
concept of exclusion-volume, and that all references used to support
ideas of “complete exclusion” rests on the Poisson-Boltzmann
equation. The Poisson-Boltzmann equation, however, predicts anions
everywhere! Thus, the concept of an anion-accessible porosity, and the
related idea that compacted bentonite contains different “types” of
water, have not been provided with any kind of theoretical support.
[1] This figure is just an illustration, not an actual result. Update (220831): Actual solutions to the Poisson-Boltzmann equation are presented here.
[2] Schofield writes with an enthusiasm seldom seen in modern scientific papers: “I considered that it would be possible to compute the negative adsorption of the repelled ions from the basic assumptions of Gouy’s theory of the diffuse electric double layer, and therefore invited Mr. M. H. Quenouille to tackle the mathematical difficulties involved. Complete solutions have now been obtained for electrolytes in which the ions have valency ratios 1:2, 1:1, and 2:1, and a full account of this work will be submitted for publication shortly.”
[3] “Bentonite” is used in the following as an abbreviation of “Bentonite and claystone”.
[4] I mean that the word “probably” as used here does not belong in a scientific text.
[5] Sciencedirect.com dates this reference to 1979. The book has a second revised edition, however, published in 1982.
[6] I use quotation marks when writing “overlap” because I think this wording gives the wrong impression in compacted clay: with an average distance between montmorillonite particles of around 1 nm, the concept of individual diffuse layers has lost its meaning.
[7] I plan to comment on “stacks” in a future blog post. Update (211027):Stacks make no sense.
[8] The volume is, however, not proportional to the Debye length, but depends exponentially on ionic strength.
[9] The “anion accessible porosity” is defined in this paper as \(\epsilon_{an} = \epsilon_{free} + \epsilon_D\cdot c_D/c_{free}\), where \(\epsilon_{free}\) is the porosity of a presumed bulk water phase in the bentonite, and \(\epsilon_D\) quantifies the volume of an arbitrarily chosen “Donnan volume” which is (Donnan) equilibrated with the “free” solution. \(c_D\) is the anion concentration in this “Donnan volume”, and \(c_{free}\) is the anion concentration in the bulk water.
[10] In this context, “interlayers” are defined as being parts of “stacks”. I really need to write about “stacks”… Update (211027):Stacks make no sense
[11] Bolt and de Haan (and others) are fond of writing that anions in very narrow confinement are “almost completely excluded” or “virtually completely excluded”, indicating that they may neglect anions in these compartments, but also that they are aware of that the equations they use never give exactly zero anion concentration. When working with soil suspensions of only a few percent solids it may be a valid approximation to neglect anions in nm-wide pores. In compacted bentonite it is not.