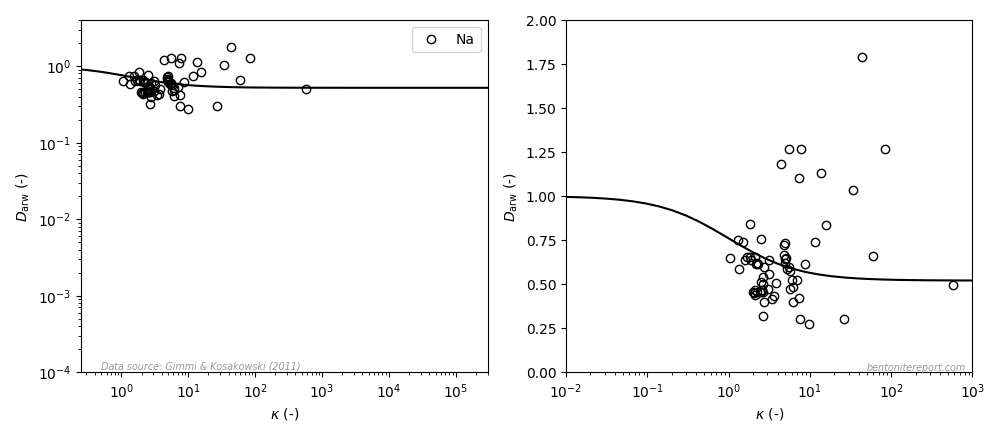
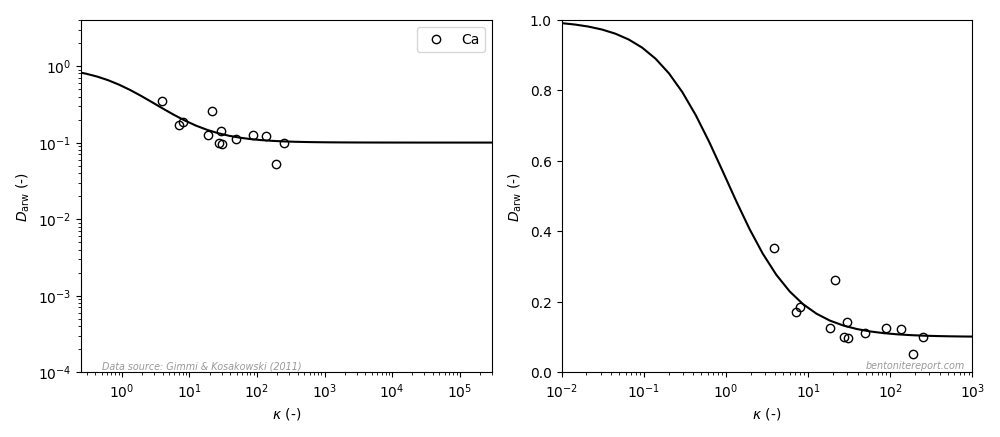
In the ongoing assessment of chloride equilibrium concentrations in bentonite, we here take a closer look at the study by Muurinen et al. (1988), in the following referred to as Mu88.1 In the quite messy plot containing all reported chloride equilibrium concentrations, we thus investigate the twelve points indicated here
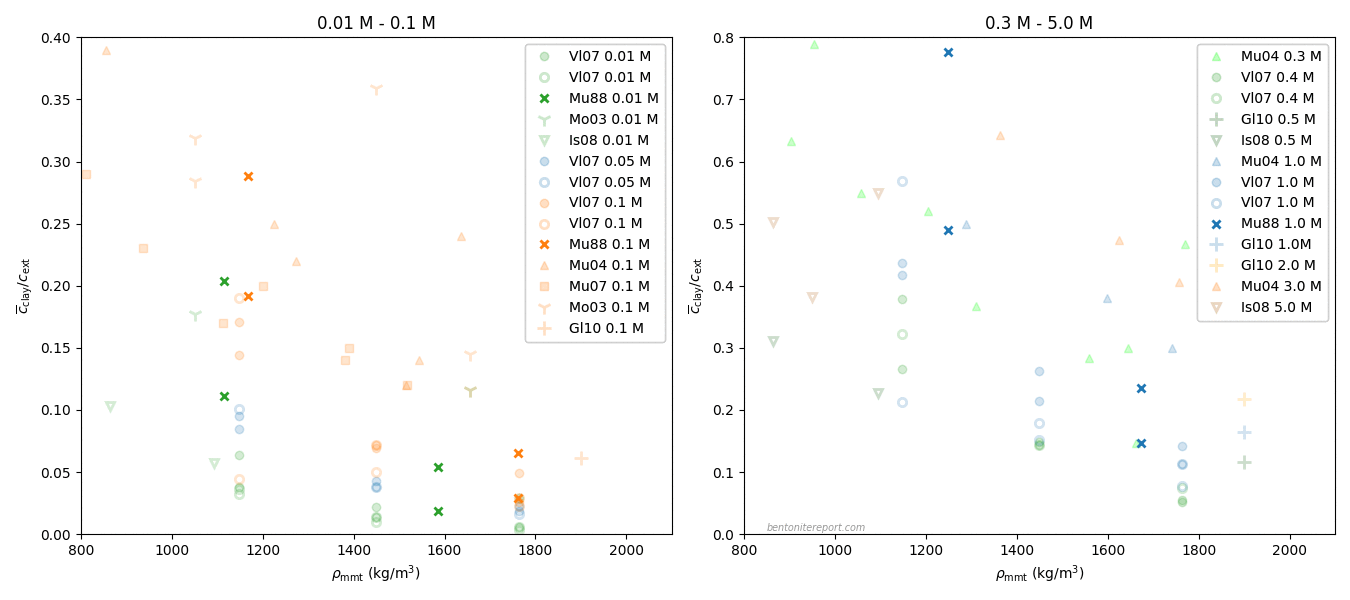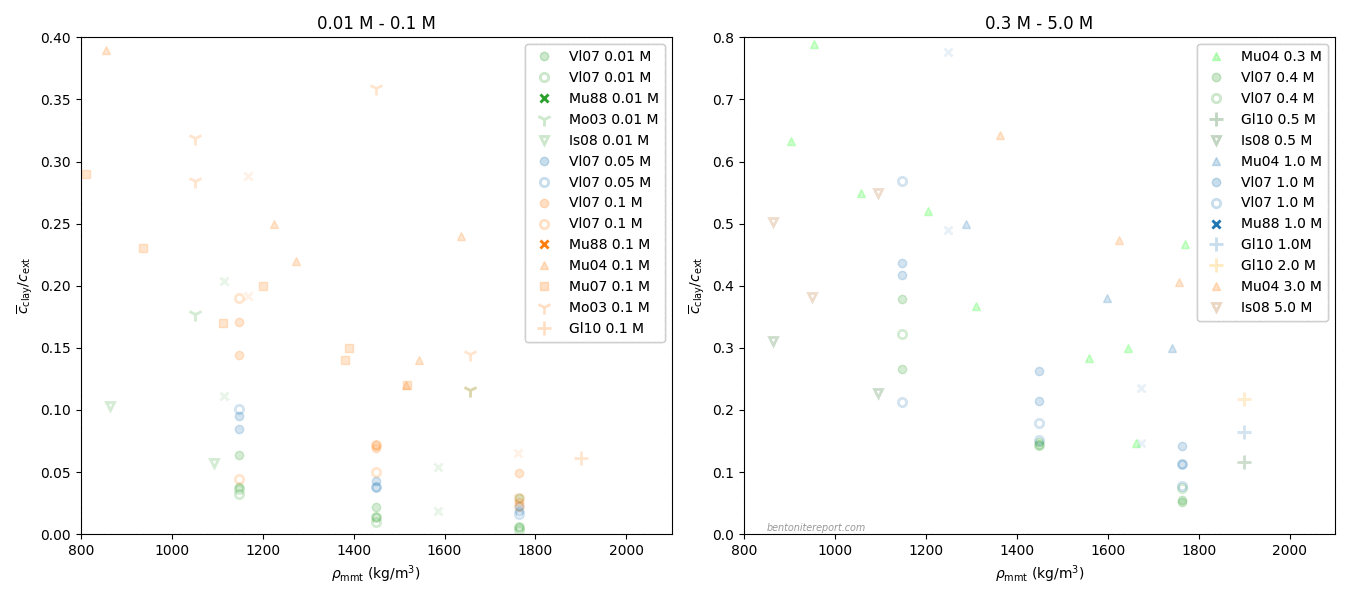
Mu88 performed both chloride and uranium through-diffusion tests on “MX-80” bentonite, as well as sorption tests. Here we focus solely on the chloride diffusion. We also disregard one diffusion test series that does not vary external concentration (it was conducted with an unspecified “artificial groundwater” and varied sample density).
Left are two test series performed with nominal sample densities 1.2 g/cm3 and 1.8 g/cm3, respectively. For each of these densities, chloride through-diffusion tests were performed with external NaCl concentrations of 0.01 M, 0.1 M, and 1.0 M, respectively. The samples were cylindrical with a diameter of 3.0 cm, and a length of 1.5 cm, giving a volume of 10.6 cm3. To refer to a specific test or sample, we use the nomenclature “nominal density/external concentration”, e.g. the test performed at nominal density 1.2 g/cm3 and external solution 0.1 M is referred to as “1.2/0.1”.
Uncertainty of bentonite samples
“MX-80” is not the name of some specific standardized material, but simply a product name.2 It is quite peculiar that that “MX-80” nevertheless is a de facto standard in the research field for clay buffers in radwaste repositories. But, being a de facto standard, several batches of bentonite with this name have been investigated and reported throughout the years. We consequently have some appreciation for its constitution, and the associated variation.
In Mu88, the material used is only mentioned by name, and it is only mentioned once (in the abstract!). We therefore can’t tell which of the studies that is more appropriate to refer to. Instead, let’s take a look at how “MX-80” has been reported generally.
| Report | Batch year | Mmt content | CEC | Na-content |
| (%) | (eq/kg) | (%) | ||
| TR-06-30 (“WySt”) | 1980 | 82.5 | 0.76 | 83 |
| NTR 83-12 | < 1983 | 75.5 | 0.76 | 86 |
| TR-06-30 (“WyL1”) | 1995 | 79.5 | 0.77 | – |
| TR-06-30 (“WyL2”) | 1999 | 79.8 | 0.75 | 71 |
| TR-06-30 (“WyR1”) | 2001 | 82.7 | 0.75 | 75 |
| TR-06-30 (“WyR2”) | 2001 | 80.0 | 0.71 | 71 |
| NTB 01-08 | < 2002 | – | 0.79* | 85 |
| WR 2004-023 | < 2004 | 80 — 85 | 0.84* | 65 |
Montmorillonite content
Reported montmorillonite content varies in the range 75 — 85%. For the present context, this primarily gives an uncertainty in adopted effective montmorillonite dry density, which, in turn, is important for making relevant comparison between bentonite materials with different montmorillonite content. For the “MX-80” used in Mu88 we here assume a montmorillonite content of 80%. In the table below is listed the corresponding effective montmorillonite densities when varying the montmorillonite content in the range \(x =\) 0.75 — 0.85, for the two nominal dry densities.
| Dry density | EMDD (\(x\)=0.75) | EMDD (\(x\)=0.80) | EMDD (\(x\)=0.85) |
| (g/cm3) | (g/cm3) | (g/cm3) | (g/cm3) |
| 1.2 | 1.01 | 1.05 | 1.09 |
| 1.8 | 1.61 | 1.66 | 1.70 |
The uncertainty in montmorillonite content thus translates to an uncertainty in effective montmorillonite dry density on the order of 0.1 g/cm3.
Cation population
While reported values of the cation exchange capacity of “MX-80” are relatively constant, of around 0.75 eq/kg,4 the reported fraction of sodium ions is seen to vary, in the range 70 — 85 %. The remaining population is mainly di-valent rare-earth metal ions (calcium and magnesium). This does not only mean that different studies on “MX-80” may give results for quite different types of systems, as the mono- to di-valent ion ratio may vary, but also that samples within the study may represent quite different systems. We examine this uncertainty below, when discussing the external solutions.
Soluble calcium minerals
The uncertainty of how much divalent cations are available is in fact larger than just discussed. “MX-80” is reported to contain a certain amount of soluble calcium minerals, in particular gypsum. These provide additional sources for divalent ions, which certainly will be involved in the chemical equilibration as the samples are water saturated. Reported values of gypsum content in “MX-80” are on the order of 1%. With a molar mass of 0.172 kg/mol, this contributes to the calcium content by \(2\cdot 0.01/0.172\) eq/kg \(\approx 0.12\) eq/kg, or about 16% of the cation exchange capacity.
Sample density
The samples in Mu88 that we focus on have nominal dry density of 1.2 and 1.8 g/cm3. The paper also reports measured porosities on each individual sample, listed in the below table together with corresponding values of dry density5
| Test | \(\phi\) | \(\rho_d\) |
| (-) | (g/cm3) | |
| 1.2/0.01 | 0.54 | 1.27 |
| 1.2/0.1 | 0.52 | 1.32 |
| 1.2/1.0 | 0.49 | 1.40 |
| 1.8/0.01 | 0.37 | 1.73 |
| 1.8/0.1 | 0.31 | 1.89 |
| 1.8/1.0 | 0.34 | 1.81 |
We note a substantial variation in measured density for samples with the same nominal density: for the 1.2 g/cm3 samples, the standard deviation is 0.06 g/cm3, and for the 1.8 g/cm3 samples it is 0.07 g/cm3. Moreover, while the mean value for the 1.8 g/cm3 samples is close to the nominal value (1.81 g/cm3), that for the 1.2 g/cm3 samples is substantially higher (1.33 g/cm3).
It is impossible to know from the information provided in Mu88 if this uncertainty is intrinsic to the procedure of preparing the samples, or if it is more related to the procedure of measuring the density at test termination.6
Uncertainty of external solutions
Mu88 do not describe how the external solutions were prepared. We assume here, however, that preparing pure NaCl solutions gives no significant uncertainty.
Further, the paper contains no information on how the samples were water saturated, nor on the external solution volumes. Since samples with an appreciable amount of di-valent cations are contacted with pure sodium solutions, it is unavoidable that an ion exchange process is initiated. As we don’t know any detail of the preparation process, this introduces an uncertainty of the exact aqueous chemistry during the course of a test.
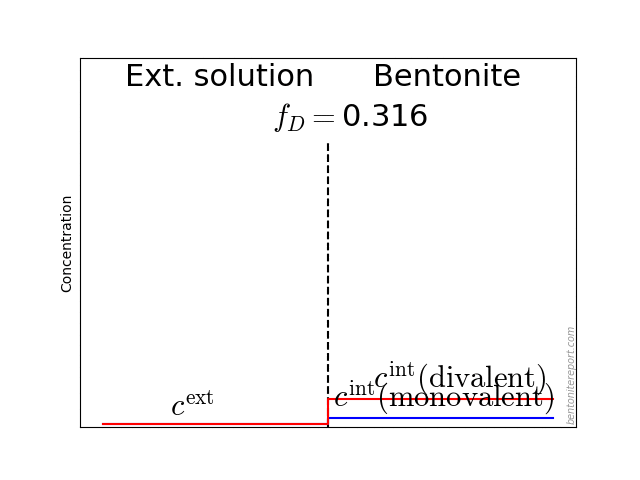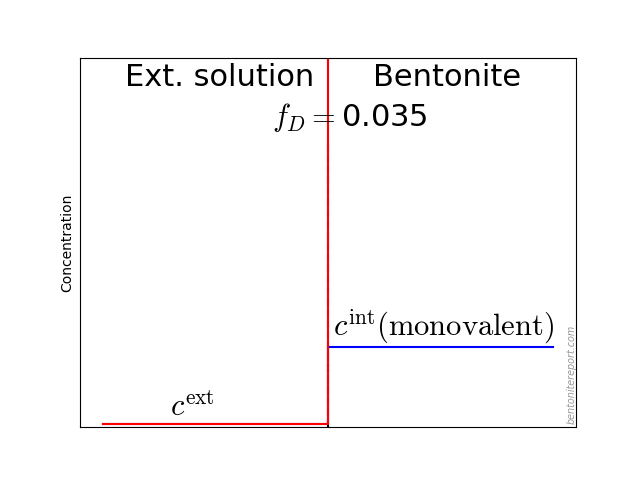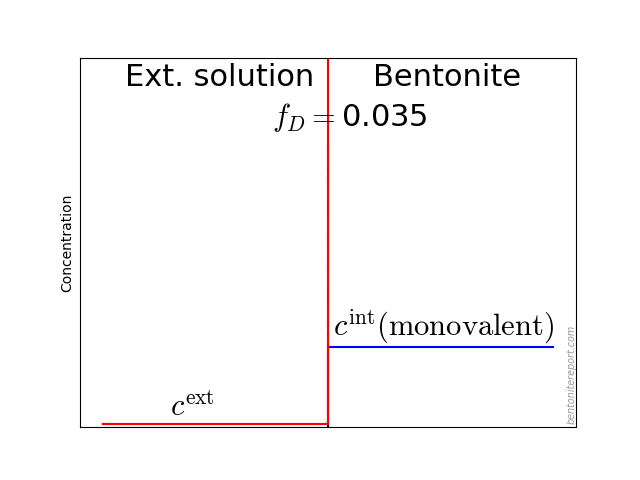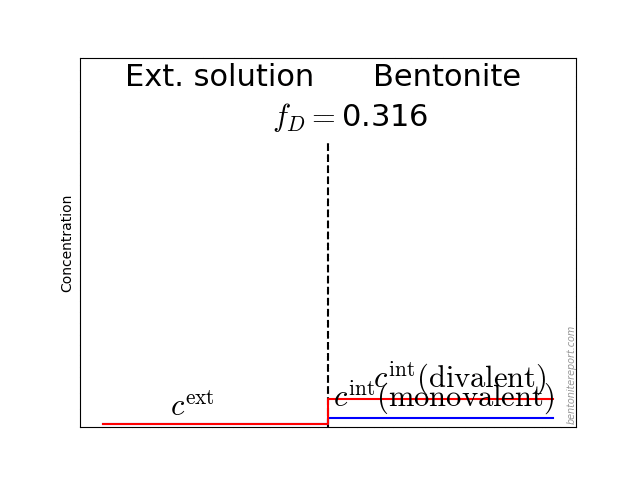
To illustrate this problem, here are the results from calculating the exchange equilibrium between a sample initially containing 30% exchangeable charge in form of calcium (70% sodium), and external NaCl solutions of various concentrations and volumes

In these calculations we assume a sample of density 1.8 g/cm3 with the same volume as in Mu88 (10.6 cm3), a cation exchange capacity of 0.75 eq/kg, and a Ca/Na selectivity coefficient of 5.
In a main series, we varied the external volume between 50 and 1000 ml (solid lines). While the solution volume naturally has a significant influence on the process, it is seen that the initial calcium content essentially remain for the lowest concentration (0.01 M). In contrast, for a 1.0 M solution, a significant amount of calcium is exchanged for all the solution volumes.
The figure also shows a case for sample density 1.2 g/cm3 (dashed line), and a scenario where equilibrium has been obtained twice, with a replacement of the first solution (to a once again pure NaCl solution) (dot-dashed line).
The main lesson from these simulations is that the actual amount of di-valent ions present during a diffusion test depends on many details: the way samples were saturated, volume of external solutions, if and how often solutions were replaced, time, etc. It is therefore impossible to state the exact ion population in any of the tests in Mu88. But, guided by the simulations, it seems very probable that the tests performed at 0.01 M contain a substantial amount of di-valent ions, while those performed at 1.0 M probably resemble more pure sodium systems.
The only information on external solutions in Mu88 is that the “solution on the low concentration side was changed regularly” during the course of a test. This implies that the amount of di-valent cations may not even be constant during the tests.
Uncertainty of diffusion parameters
The diffusion parameters explicitly listed in Mu88 are \(D_e\) and “\(D_a\)”, while it is implicitly understood that they have been obtained by fitting the effective porosity model to outflux data and the measured clay concentration profile in the final state. “\(D_a\)” is thus really the pore diffusivity \(D_p\),7 and relates to \(D_e\) as \(D_e = \epsilon_\mathrm{eff} D_p\), where \(\epsilon_\mathrm{eff}\) is the so-called “effective porosity”. In a previous blog post, we discussed in detail how anion equilibrium concentrations can be extracted from through-diffusion tests, and the results derived there is used extensively in this section.
Rather than fitting the model to the full set of data (i.e. outflux evolution and final state concentration profile), diffusion parameters in Mu88 have been extracted in various limits.
Evaluation of \(D_e\) in Mu88
The effective diffusivity was obtained by estimating the steady-state flux, dividing by external concentration difference of the tracer, and multiplying by sample length \begin{equation} D_e = \frac{j^\mathrm{ss}\cdot L}{c^\mathrm{source}}\tag{1} \end{equation}
Here it is assumed that the target reservoir tracer concentration can be neglected (we assume this throughout). Eq. 1 is basically eq. 1 in Mu88 (and eq. 8 in the earlier blog post), from which we can evaluate the values of the steady-state flux that was used for the reported values of \(D_e\) (\(A \approx 7.1\) cm2 denotes sample cross sectional area)
| Test | \(D_e\) | \(A\cdot j^\mathrm{ss}/c^\mathrm{source}\) |
| (\(\mathrm{m^2/s}\)) | (ml/day) | |
| 1.2/0.01 | \(7.7\cdot 10^{-12}\) | 0.031 |
| 1.2/0.1 | \(2.9\cdot 10^{-11}\) | 0.118 |
| 1.2/1.0 | \(1.2\cdot 10^{-10}\) | 0.489 |
| 1.8/0.01 | \(3.3\cdot 10^{-13}\) | 0.001 |
| 1.8/0.1 | \(4.8\cdot 10^{-13}\) | 0.002 |
| 1.8/1.0 | \(4.0\cdot 10^{-12}\) | 0.016 |
The figure below compares the evaluated values of the steady-state flux with the flux evaluated from the measured target concentration evolution,8 for samples with nominal dry density 1.8 g/cm3 (no concentration data was reported for the 1.2 g/cm3 samples)
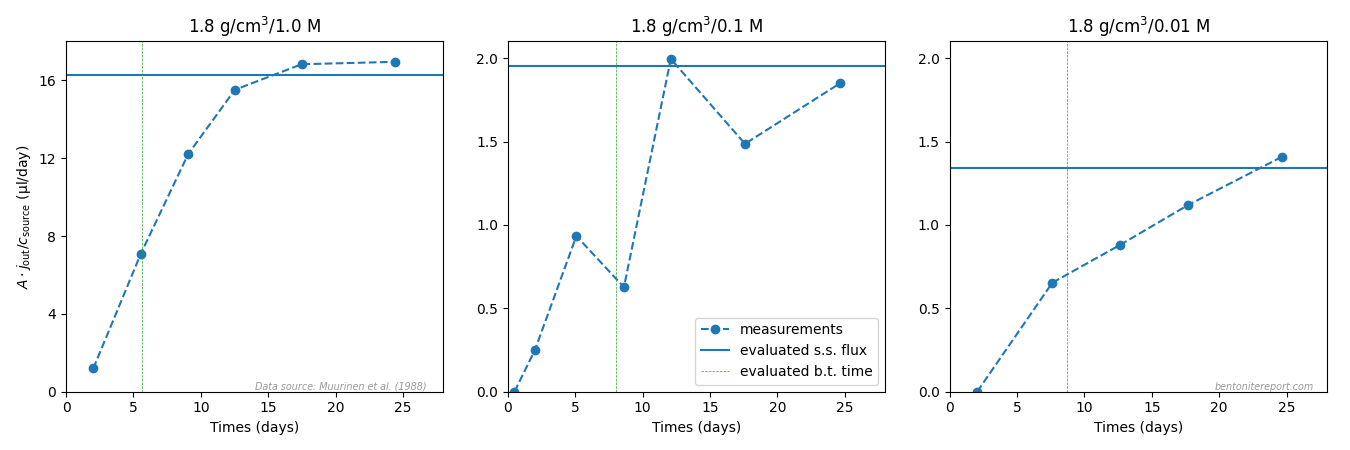
These plots clearly show that the transition to steady-state is only resolved properly for the test with highest background concentration (1.0 M). It follows that the uncertainty of the evaluated steady-state — and, consequently, of the evaluated \(D_e\) values — increases dramatically with decreasing background concentration for these samples.
Evaluation of \(D_p\) in Mu88
Pore diffusivities were obtained in two different ways. One method was to relate the steady-state flux to the clay concentration profile at the end of the test, giving \begin{equation} D_{p,c} = \frac{j^\mathrm{ss}\cdot L}{\phi\cdot\bar{c}(0)} \tag{2} \end{equation}
where \(\bar{c}(0)\) denotes the chloride clay concentration at the interface to the source reservoir. The quantity in eq. 2 is called “\(D_{ac}\)”7 in Mu88, and this equation is essentially the same as eq. 2 in Mu889 (and eq. 10 in the previous blog post). Using the steady-state fluxes, we can back-calculate the values of \(\bar{c}(0)\) used for this evaluation of \(D_{p,c}\)
| Test | \(D_{p,c}\) | \(A\cdot j^\mathrm{ss}/c^\mathrm{source}\) | \(\phi\) | \(\bar{c}(0)/c^\mathrm{source}\) |
| (\(\mathrm{m^2/s}\)) | (ml/day) | (-) | (-) | |
| 1.2/0.01 | \(7.0\cdot 10^{-11}\) | 0.031 | 0.54 | 0.204 |
| 1.2/0.1 | \(2.8\cdot 10^{-10}\) | 0.118 | 0.52 | 0.199 |
| 1.2/1.0 | \(5.1\cdot 10^{-10}\) | 0.489 | 0.49 | 0.480 |
| 1.8/0.01 | \(2.0\cdot 10^{-11}\) | 0.001 | 0.37 | 0.045 |
| 1.8/0.1 | \(3.1\cdot 10^{-11}\) | 0.002 | 0.31 | 0.050 |
| 1.8/1.0 | \(5.2\cdot 10^{-11}\) | 0.016 | 0.34 | 0.226 |
Note that, although we did some calculations to obtain them, the values for \(\bar{c}(0)/c^\mathrm{source}\) in this table are closer to the actual measured raw data (concentrations). We made the calculation above to “de-derive” these values from the reported diffusion coefficients (combining eqs. 1 and 2 shows that \(\bar{c}(0)\) is obtained from the reported parameters as \(\bar{c}(0)/c^\mathrm{source} = D_e/(\phi D_{p,c})\)).
Here are compared the measured concentration profiles for the samples of nominal density 1.8 g/cm3 and the corresponding slopes used to evaluate \(D_{p,c}\) (profiles for the 1.2 g/cm3 samples are not provided in Mu88)

For background concentrations 1.0 M and 0.1 M, the evaluated slope corresponds quite well to the raw data. For the 0.01 M sample, however, the match not very satisfactory. I suspect that a detection limit may have been reached for the analysis of the profile of this sample. Needless to say, the evaluated value of \(\bar{c}(0)\) is very uncertain for the 0.01 M sample.
It may also be noted that all measured concentration profiles deviates from linearity near the interface to the source reservoir. This is a general behavior in through-diffusion tests, which I am quite convinced of is related to sample swelling during dismantling, but there are also other suggested explanations. Here we neglect this effect and relate diffusion quantities to the linear parts of profiles, but this issue should certainly be treated in a separate discussion. Update (220407): non-linear profiles are discussed here.
\(D_p\) was also evaluated in a different way in Mu88, by measuring what we here will call the breakthrough time, \(t_\mathrm{bt}\) (Mu88 call it “time-lag”). This quantity is fairly abstract, and relates to the asymptotic behavior of the analytical expression for the outflux that apply for constant boundary concentrations (we here assume them to be \(c^\mathrm{source}\) and 0, respectively). This expression is displayed in eq. 7 in the previous blog post.
Multiplying the outflux by the sample cross sectional area \(A\) and integrating, gives the accumulated amount of diffused tracers. In the limit of long times, this quantity is, not surprisingly, linear in \(t\) \begin{equation} A\cdot j^\mathrm{ss} \cdot \left(t – \frac{L^2}{6\cdot D_p} \right ) \end{equation}
\(t_\mathrm{bt}\) is defined as the time for which this asymptotic expression is zero. Determining \(t_\mathrm{bt}\) from the measured outflux evolution consequently allows for an estimation of \(D_p\) as \begin{equation} D_{p,t} = \frac{L^2}{6t_\mathrm{bt}} \tag{3} \end{equation}
This quantity is called “\(D_{at}\)” in Mu887 (eq. 3 is eq. 3 in Mu88). With another back calculation we can extract the values of \(t_\mathrm{bt}\) determined from the raw data
| Test | \(D_{p,t}\) | \(t_\mathrm{bt}\) |
| (\(\mathrm{m^2/s}\)) | (days) | |
| 1.2/0.01 | \(1.4\cdot 10^{-10}\) | 3.1 |
| 1.2/0.1 | \(2.0\cdot 10^{-10}\) | 2.2 |
| 1.2/1.0 | \(3.2\cdot 10^{-10}\) | 1.4 |
| 1.8/0.01 | \(5.0\cdot 10^{-11}\) | 8.7 |
| 1.8/0.1 | \(5.4\cdot 10^{-11}\) | 8.0 |
| 1.8/1.0 | \(7.7\cdot 10^{-11}\) | 5.6 |
These evaluated breakthrough times are indicated in the flux plots above for samples of nominal dry density 1.8 g/cm3. For the 0.1 M and 0.01 M samples it is obvious that this value is very uncertain — without a certain steady-state flux it is impossible to achieve a certain breakthrough time. The breakthrough time for the 1.8/1.0 test, on the other hand, simply appears to be incorrectly evaluated: in terms of outflux vs. time, the breakthrough time should be the time where the flux has reached 62% of the steady-state value.10
As no raw data is reported for the 1.2 g/cm3 tests, the quality of the evaluated breakthrough times cannot be checked for them. It may be noted, however, that the evaluated breakthrough times are significantly shorter in this case as compared with the 1.8 g/cm3 tests. Consequently, while the sampling frequency is high enough to properly resolve the transient stage of the outflux evolution for the 1.8g/cm3 tests, it must be substantially higher in order to resolve this stage in the 1.2g/cm3 tests (I guess a rule of thumb is that sampling frequency must be at least higher than \(1/t_{bt}\)).
With the pore diffusivities evaluated from \(t_\mathrm{bt}\) we get a second estimation of \(\bar{c}(0)/c^\mathrm{source}\), using eq. 2. These values are listed in the table below and compared with the direct evaluation from the steady-state concentration profiles.
| Test | \(\bar{c}(0)/c^\mathrm{source}\) | \(\bar{c}(0)/c^\mathrm{source}\) |
| (breakthrough) | (profile) | |
| 1.2/0.01 | 0.102 | 0.204 |
| 1.2/0.1 | 0.279 | 0.199 |
| 1.2/1.0 | 0.765 | 0.480 |
| 1.8/0.01 | 0.018 | 0.045 |
| 1.8/0.1 | 0.029 | 0.050 |
| 1.8/1.0 | 0.153 | 0.226 |
In a well conducted study these estimates should be similar; \(D_{p,c}\) and \(D_{p,t}\) are, after all, estimations of the same quantity: the pore diffusivity \(D_p\).7 But here we note a discrepancy of approximately a factor 2 between several values of \(\bar{c}(0)\).
It is difficult to judge generally which of the estimations are more accurate, but we have seen that for the 1.8/0.1 and 1.8/0.01 tests, the flux data is not very well resolved, giving a corresponding uncertainty on the equilibrium concentration estimated from the breakthrough time. On the other hand, also the concentration profile is poorly resolved in the case of 0.01 M at 1.8 g/cm3.
However, in cases where the value of \(\bar{c}(0)/c^\mathrm{source}\) is substantial (as for the 1.8/1.0 test and, reasonably, for all tests at 1.2 g/cm3), we expect the estimation directly from the concentration profile to be accurate and robust (as for the 1.8 g/cm3 test at high NaCl concentration). For the 1.2 g/cm3 samples we cannot say much more than this, since Mu88 don’t provide the concentration raw data. For the 1.8/1.0 test, however, we can continue the analysis by fitting the model to all available data.
Re-evaluation by fitting to the full data set
Note that all evaluations in Mu88 are based on making an initial estimation of the steady-state flux, giving \(D_e\) (eq. 1). This value of \(D_e\) (or \(j^{ss}\)) is thereafter fixed in the subsequent estimation of \(D_{p,c}\) (eq. 2). Likewise, an estimation of the steady-state flux is required for estimating the breakthrough time. Here is an animation showing the variation of the model when transitioning from the value of the pore diffusivity estimated from breakthrough time (\(7.7\cdot 10^{-11}\) m2/s), to the value estimated from concentration profile (\(5.2\cdot 10^{-11}\) m2/s) for the 1.8/1.0 test, keeping the steady-state flux fixed at the initial estimation

Note that the axes for the flux is on top (time) and to the right (accumulation rate). This animation confirms that the diffusivity evaluated from breakthrough time in Mu88 gives a way too fast process: the slope of the steady-state concentration profile is too small, and the outflux evolution has a too short transient stage. On the other hand, using the diffusivity estimated from the concentration profiles still doesn’t give a flux that fit very well. The problem is that this fitting is performed with a fixed value of the steady-state flux. By instead keeping the slope fixed at the experimental values, while varying diffusivity (and thus steady state flux), we get the following variation

This animation shows that the model can be fitted well to all data (at least for the 1.8/1.0 test). The problem with the evaluation in Mu88 is that it assumes the steady-state to be fully reached at the later stages of the test. As the above fitting procedure shows, this is only barely true. The experiments could thus have been designed better by conducting them longer, in order to better sample the steady-state phase (and the steady-state flux should have been fitted to the entire data set). Nevertheless, for this sample, the steady-state flux obtained by allowing for this parameter to vary is only slightly different from that used in Mu88 (17.5 rather than 16.3 \(\mathrm{\mu}\)l/day, corresponding to a change of \(D_p\) from \(5.2\cdot10^{-11}\) to \(5.6\cdot10^{-11}\) m2/s). Moreover, this consideration should not be a problem for the 1.2 g/cm3 tests, if they were conducted for as long time as the 1.8 g/cm3 tests, because steady-state is reached much faster (in those tests, sampling frequency may instead be a problem, as discussed above).
As we were able to fit the full model to all data, we conclude that the value of \(\bar{c}(0)/c^\mathrm{source}\) obtained from \(D_{p,c}\) is probably the more robust estimation11, and that there appears to be a problem with how the breakthrough times have been determined. For the 1.8 g/cm3 samples we have demonstrated that this is the case, for the 1.2 g/cm3 we can only make an educated guess that this is the case.
Summary and verdict
We have seen that the results on chloride diffusion in Mu88 suffer from uncertainty from several sources:
- The “MX-80” material is not that well defined
- Densities vary substantially for samples at the same nominal density
- Without knowledge of e.g water saturation procedures and solution volumes, it is impossible to estimate the proper ion population during the course of a test
- It is, however, highly likely that tests performed at low NaCl concentrations contain substantial amounts of di-valent ions, while those at high NaCl concentration are closer to being pure sodium systems.
- The reported diffusivities give a corresponding uncertainty in the chloride equilibrium concentrations of about a factor of 2. While some tests essentially have a too high noise level to give certain estimations, the problem for the others seems to stem from the estimation of breakthrough times.
Here is an attempt to encapsulate the above information in an updated plot for the chloride equilibrium data in Mu88

The colored squares represent “confidence areas” based on the variation within each nominal density (horizontally), and on the variation of \(\bar{c}(0)/c^\mathrm{source}\) from the two reported values on pore diffusivity7 (vertically). The limits of these rectangles are simply the 95% confidence interval, based on these variations, and assuming a normal distribution.
Data points put within parentheses are estimations judged to be improper (based on either re-evaluation of the raw data, or informed guesses).
From the present analysis my decision is to not use the data from Mu88 to e.g. validate models for anion exclusion. Although there seems to be nothing fundamentally wrong with how these test were conducted, they suffer from so many uncertainties of various sources that I judge the data to not contribute to quantitative process understanding.
Footnotes
[1] This work is referred to as “Muurinen et al. (1989)” by several authors.
[2] MX-80 is not only a brand name, but also a band name.
[3] This report is “Bentonite Mineralogy” by L. Carlson (Posiva WR 2004-02), but it appears to not be included in the INIS database. It can, however, be found with some elementary web searching.
[4] It’s interesting to note that the cation exchange capacity of “MX-80” remains more or less constant, while the montmorillonite content has some variation. This implies that the montmorillonite layer charge varies (and is negatively correlated with montmorillonite content). Could it be that the manufacturer has a specified cation exchange capacity as requirement for this product?
[5] To convert porosity to dry density, I used \(\rho_d = \rho_s\cdot(1-\phi)\), with solid grain density \(\rho_s = 2.75\) g/cm3.
[6] A speculation is that the uncertainty stems from the measurement procedure, as this was done on smaller sections of the full samples. It is not specified in Mu88 what the reported porosity represent, but it is reasonable to assume that it is the average of all sections of a sample.
[7] At the risk of losing some clarity, I refuse to use the term “apparent diffusivity” for something which actually is a real pore diffusivity.
[8] These values were not tabulated, but I have read them off from the graphs in Mu88.
[9] Mu88 use the concentration based on the total volume in their expression, while \(\bar{c}\) is defined in terms of water volume (water mass, strictly). Eq.2 therefore contains the physical porosity. In their concentration profile plots, however, Mu88 use \(\bar{c}\) as variable (called \(c_{pw}\) — the “concentration in the pore water”)
[10] Plugging the breakthrough time \(L^2/6D_p\) into the expression for the flux gives
\begin{equation} j^\mathrm{out}(t_\mathrm{bt}) = j^{ss}\cdot\left ( 1 + 2 \sum (-1)^n e^\frac{-\pi^2 n^2}{6} \right ) \approx 0.616725\cdot j^{ss} \end{equation}
I find it amusing that this value is close to the reciprocal golden ratio (0.618033…). Finding the breakthrough time from a flux vs. time plot thus corresponds (approximately) to splitting the y-axis according to the golden ratio.
[11] Note that the actual evaluated values of $D_{p,c}$ in Mu88 still may be uncertain, because they also depend on the values of the steady-state flux, which we have seen were not optimally evaluated.