There are several “descriptions” of bentonite swelling. While a few of them actually denies any significant role played by the exchangeable cations, most of these descriptions treat the exchangeable ions as part of an osmotic system. I have earlier discussed how the terms “osmotic” or “osmosis” may cause some confusion in different contexts, and discussed the confusion surrounding the treatment of electrostatic forces.
In this blog post I discuss the description of bentonite swelling
often adopted in the fields of soil mechanics and geotechnical
engineering. In particular, we focus on the concept of suction,
which is central in these research fields, while being basically
absent in others.
As far as I understand, suction is just the water chemical potential “disguised” as a pressure variable; although I have trouble finding clear-cut definitions, it seems clear that suction is directly inherited from the “water potential” concept, which has been central in soil science for a long time. Applied to bentonite, the geotechnical description is thus not principally different from the osmotic approach that I have presented previously. But the way the suction concept is (and isn’t) applied may cause unnecessary confusion regarding the swelling mechanisms. I think a root for this confusion is that suction involves both osmotic and capillary mechanisms.
Matric suction (capillary suction)
Matric suction is typically associated with capillarity, a fundamental mechanism in many conventional soil materials under so-called unsaturated conditions. A conventional soil with a significant amount of small enough pores shows capillary condensation, i.e. it contains liquid water below the condensation point for ordinary bulk water. Naturally, the equilibrium vapor pressure increases with the amount of water in the soil, as the pores containing liquid water become larger. For conventional soils, it therefore makes sense to speak of the degree of saturation of a sample, and to relate saturation and equilibrium vapor pressure by means of a water retention curve. Underlying this picture is the notion that the solid parts constitute a “soil skeleton” (the matrix), and that the soil can be viewed as a vessel that can be more or less filled with water.
The pressure of the capillary water is lower than that of the surrounding air, and is related to the curvature of the interfaces between the two phases (menisci), as expressed by the Young-Laplace equation. For a spherically symmetric meniscus this equation reads
\begin{equation} \Delta p = p_w – p_a = \frac{2\sigma}{r} \tag{1} \end{equation}
where \(p_a\) and \(p_w\) denote the pressures of air and capillary water,
respectively, \(\sigma\) is the surface tension, and \(r\) is the radius
of curvature of the interface. The sign of \(r\) depends on whether the
interface bulges inwards (“concave”, \(r<0\)) or outwards (“convex”,
\(r>0\)). For capillary water, \(r\) is negative and \(\Delta p\) — which
is also called the
Laplace
pressure — is a negative quantity.
As far as I understand, matric suction is simply defined as the negative Laplace pressure, i.e.
\begin{equation} s_m = p_a – p_w \tag{2} \end{equation}
With this definition, suction has a straightforward physical meaning
as quantifying the difference in pressure of the two fluids occupying
the pore space, and clearly relates to the everyday use of the word.
Suction — in this capillary sense — gives a simple principal
explanation for (apparent) cohesion in e.g. unsaturated sand:
individual grains are pushed together by the air-water pressure
difference, as schematically illustrated here (the yellow stuff is
supposed to be two grains of sand, and the blue stuff water)
It is reasonable to assume that the net force transmitted by the soil skeleton — usually quantified using the concept of effective stress — governs several mechanical properties of the soil sample, e.g. shear strength. The above description also makes it reasonable to assume that effective stress depends on suction.
Thus, in unsaturated conventional soil are quantities like degree of saturation, pore size distribution, (matric) suction, effective stress, and shear strength very much associated. Another way of saying this is that there is an optimal combination of water content and particle size distribution for constructing the perfect sand castle.
The chemical potential of the capillary water is related to matric suction. Choosing pure bulk water under pressure \(p_a\)1 as reference, the chemical potential of the liquid phase in the soil is obtained by integrating the Gibbs-Duhem equation from \(p_a\) to \(p_w\)
\begin{equation} \mu = \mu_0 + \int_{p_a}^{p_w}v dP = \mu_0 + v\cdot \left(p_w – p_a \right) = \mu_0 -v\cdot s_m \tag{3} \end{equation}
where \(\mu_0\) is the reference chemical potential, \(v\) is the molar volume of water, and we have assumed incompressibility.
The above expression shows that matric suction in this case directly quantifies the (relative) water chemical potential. Note, however, that eq. 3 does not define matric suction; \(s_m\) is defined as a pressure difference between two phases (eq. 2), and happens to quantify the chemical potential under the present circumstances (pure capillary water).
A chemical potential can generally be expressed in terms of activity (\(a\))
\begin{equation} \mu = \mu_0 + RT \ln a \tag{4} \end{equation}
For our case, water activity is to a very good approximation equal to relative humidity, the ratio between the vapor pressures in the state under consideration and in the reference state, i.e. \(a = p_v/p_{v,0}\). Combining eqs. 3 and 4, we see that the vapor pressure in this case is related to matric suction as
\begin{equation} \frac{p_v}{p_{v,0}} = e^{-v\cdot s_m/RT} \end{equation}
Using the Young-Laplace equation (eq. 1) for \(s_m\) we can also write this as
\begin{equation} \frac{p_v}{p_{v,0}} = e^{\frac{2v\sigma}{RTr}} \end{equation}
This is the so-called
Kelvin
equation, which relates the equilibrium vapor pressure to the
curvature of an air-pure water interface. Note that, since \(r<0\) for
capillary water, the vapor pressure is lower than the corresponding
bulk value (\(p_v < p_{v,0}\)).
Osmotic suction and total suction
So far, we have discussed suction in a capillary context, and related it to water chemical potential or vapor pressure. Now consider how the picture changes if the pores in our conventional soil contain saline water. Matric suction — i.e. the actual pressure difference between the pore solution and the surrounding air, sticking with eq. 2 as the definition — is in general different from the pure water case, because solutes influence surface tension. Also, water activity (vapor pressure) is different from the pure water case, but there is no longer a direct relation between water activity and matric suction, because water activity is independently altered by the presence of solutes.
The water chemical potential of a saline bulk solution (i.e. with no capillary effects), can be written in terms of the osmotic pressure, \(\pi(c)\)
\begin{equation} \mu(c) = \mu_0 – v\cdot\pi(c) \tag{5} \end{equation}
where we have assumed a salt concentration \(c\), and indicated that the
osmotic pressure, and hence the chemical potential, depends on this
concentration.
Although eq. 5 is of the same form as eq. 3, matric suction and osmotic pressure are very different quantities. The former is defined under circumstances where an actual pressure difference prevail between the air and water phases. In contrast, there is no pressure difference between the phases in a container containing both a solution and a gas phase. \(\pi(c)\) corresponds to the elevated pressure that must be applied for the solution to be in equilibrium with pure water kept at the reference pressure.
Despite the different natures of matric suction and osmotic pressure, the fields of geotechnical engineering and soil mechanics insist on also referring to \(\pi(c)\) as a suction variable: the osmotic suction. Similarly, total suction is defined as the sum of matric and osmotic suction
\begin{equation} \Psi = s_m + \pi(c) \end{equation}
These definitions seem to have no other purpose than to be able to write the water chemical potential generally as
\begin{equation} \mu = \mu_0 -v\cdot \Psi \tag{6} \end{equation}
Total suction is thus de facto defined simply as the (relative)
value of the water chemical potential, expressed as a pressure (I
think this is completely analogous to “total water potential” in
soil science).
Eq. 6 shows that \(\Psi\) is directly related to water activity, or vapor pressure, and we can write
\begin{equation} \frac{p_v}{p_{v,0}} = e^{-v\cdot \Psi/RT} \tag{7} \end{equation}
This relation is quite often erroneously referred to as the Kelvin equation (or “Kelvin’s law”) in the bentonite literature. But note that the above equation just restates the definition of water activity, because \(v\cdot\Psi\) cannot be reduced to anything more concrete than the relative value of the water chemical potential. The Kelvin equation, on the other hand, expresses something more concrete: the equilibrium vapor pressure for a curved air-water interface. Some clay literature refer to the above relation as the “Psychrometric law”, but that name seems not established in other fields.2
A definition is motivated by its usefulness, and total change in water chemical potential is of course central when considering e.g. moisture movement in soil. My non-geotechnical brain, however, is not fond of extending the “suction” variable in the way outlined above. To start with, there is already a variable to use: the water chemical potential. Also, “total suction” no longer has the direct relation to the everyday use of the word suction: there is no “sucking” going on in a saline bulk solution,3 while in a capillary there is. Furthermore, with a saline pore solution there is no direct relation between (total) suction and e.g. effective stress or shear strength.
Although both matric suction and osmotic pressure under certain circumstances can be measured in a direct way, it seems that (total) suction usually is quantified by measuring/controlling the vapor pressure with which the soil sample is in equilibrium. Actually, one of the more comprehensive definitions of various “suctions” that I have been able to find — in Fredlund et al. (2012) — speaks only of various vapor pressures (although based on the capillary and osmotic concepts):4
Matric or capillary component of free energy: Matric suction is the equivalent suction derived from the measurement of the partial pressure of the water vapor in equilibrium with the soil-water relative to the partial pressure of the water vapor in equilibrium with a solution identical in composition with the soil-water.
Osmotic (or solute) component of free energy: Osmotic suction is the equivalent suction derived from the measurement of the partial pressure of the water vapor in equilibrium with a solution identical in composition with the soil-water relative to the partial pressure of water vapor in equilibrium with free pure water.
Total suction or free energy of soil-water: Total suction is the equivalent suction derived from the measurement of the partial pressure of the water vapor in equilibrium with the soil-water relative to the partial pressure of water vapor in equilibrium with free pure water.
It seems that such operational definitions of suction has made
the term synonymous with “vapor pressure depression” in large parts
of the bentonite
scientific
literature.
Suction in bentonite
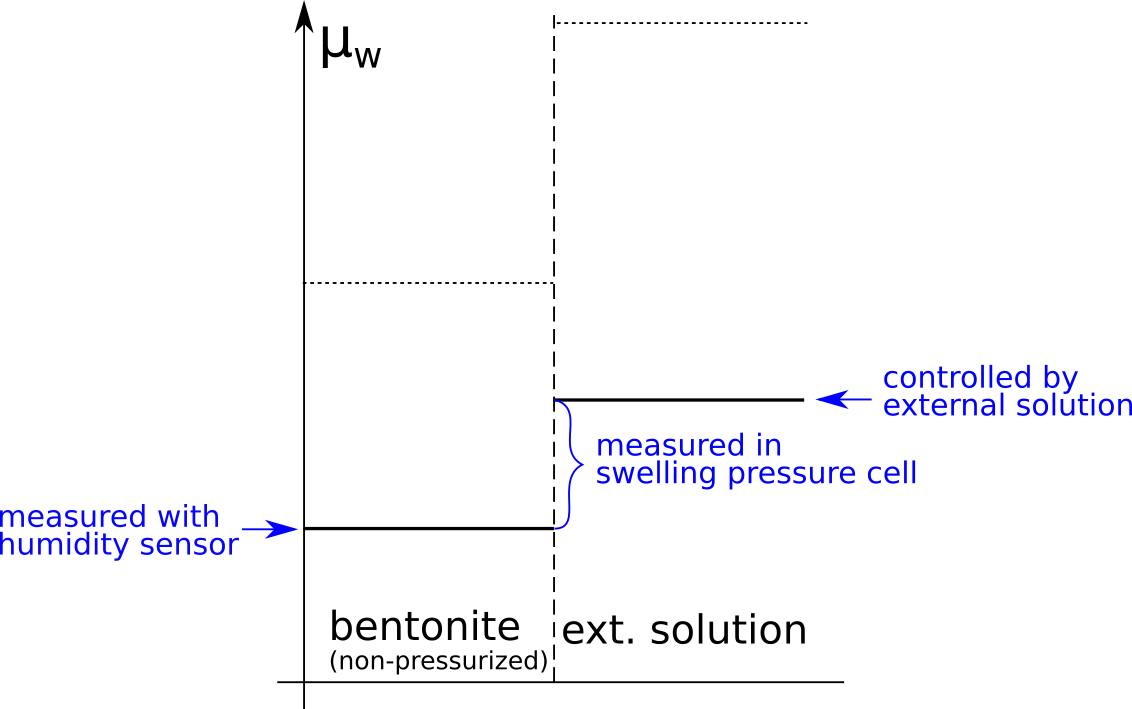
In the above discussion we had mainly a conventional soil in mind. When applying the suction concepts to bentonite,5 I think there are a few additional pitfalls/sources for confusion. Firstly, note that the definitions discussed previously involve “a solution identical in composition with the soil-water”. But soil-water that contains appreciable amounts of exchangeable ions — as is the case for bentonite — cannot be realized as an external solution.
It seems that this “complication” is treated by assuming that an external solution in equilibrium with a bentonite sample is the soil-water (this is analogous to how many geochemists use the term “porewater” in bentonite contexts). Not surprisingly, this treatment has bizarre consequences. The conclusion for e.g. a salt free bentonite sample — which is in equilibrium with pure water — is that it lacks osmotic suction, and that its lowered vapor pressure (when isolated and unloaded) is completely due to matric suction! I think this is such an odd outcome that it is worth repeating: A system dominated by interlayer pores, containing dissolved cations at very high concentrations, is described as lacking osmotic pressure! It is not uncommon to find descriptions like this one (from Lang et al. (2019))
The total suction of unsaturated soils consists of matric and
osmotic suctions (Yong and Warkentin, 1975; Fredlund et al., 2012;
Lu and Likos, 2004). In clays, the matric suction is due to surface
tension, adsorptive forces and osmotic forces (i.e. the diffuse
double layer forces), whereas the osmotic suction is due to the
presence of dissolved solutes in the pore water.
We apparently live in a world where “matric suction” consists of “osmotic forces”, while the same “osmotic forces” do not contribute to “osmotic suction”. Except when the clay contains excess ions, in which case we have an arbitrary combination of the two “suctions” (note also that “osmotic suction” and “osmotic swelling” are two quite different things).
Although the above consequence is odd, it is still only a matter of definition: accepting that “matric suction” involves osmotic forces (which I don’t recommend), the description may still be adequate in principle; after all, “total suction” quantifies the reduction of the water chemical potential.
But the focus on “matric suction” also reveals a conceptual view of bentonite structure that I find problematic: it suggests a first order approximation of bentonite as a conventional soil, i.e. as an assemblage of solid grains separated from an aqueous phase (and a gas phase). This “matric” view is fully in line with the idea of “free water” in bentonite, and it is quite clear that this is a prevailing view in the geotechnical, as well as in the geochemical, literature. For instance, with the formulation “the presence of dissolved solutes in the pore water” in the above quotation, the “pore water” the authors have in mind is a charge neutral bulk water solution.
With the “matric” conceptual view, the degree of saturation becomes a central variable in much soil mechanical analyses of bentonite. When dealing with actual unsaturated bentonite samples, I guess this makes sense, but once a sample is saturated this variable has lost much of its meaning.6 Consider e.g. the different expected behaviors if drying e.g. a water saturated metal filter or a saturated bentonite sample.
The equilibrium vapor pressure of both these systems is lower than the
corresponding pure bulk water value. For the metal filter, the lowered
water activity is of course due to capillarity, i.e. there is an
actual pressure reduction in the water phase (matric suction!). When
lowering the external vapor pressure below the equilibrium point
(i.e. drying), capillary water migrates out of the filter, while the
metal structure itself remains intact. In this case, as the system
remains defined in a reasonable way, it is motivated to speak of the
saturation state of the filter.
For a drying bentonite sample, the behavior is not as well defined,
and depends on how the drying is performed and on initial water
content. For a quasi-static process, where the external vapor pressure
is lowered in small steps at an arbitrary slow rate, it should be
clear that the entire sample will respond simply by shrinking. In
this case it does not make much sense to speak of the sample as still
being saturated, nor to speak of it as having become unsaturated.
For a more “violent” drying process, e.g. placing the bentonite sample in an oven at 105 °C , it is also clear that — rather than resulting in a neatly shrunken, dense piece of clay — the sample now will suffer from macroscopic cracks and other deformations. Neither in this case does it make much sense to try to define the degree of saturation, in relation to the sample initially put in the oven.
Note also that if we, instead of drying, increase the external
vapor pressure from the initial equilibrium value, the metal filter
will not respond much at all, while the bentonite sample immediately
will begin to swell.
I hope that this example has made it clear, not only that the degree of saturation is in general ill-defined for bentonite, but also that a bentonite sample behaves more as an aqueous solution rather than as a conventional soil: if we alter external vapor pressure, an aqueous solution responds by either “swelling” (taking up water) or “shrinking” (giving off water). A main aspect of this conceptual view of bentonite — which we may call the “osmotic” view — is that water does not form a separate phase7. This was pointed out e.g. by Bolt and Miller (1958) (referring to this type of system as an “ideal clay-water system”)
In contrast to the familiar case described is the ideal clay-water
system in which the particles are not in direct contact but are
separated by layers of water. Removal of water from such a system
does not introduce a third phase but merely causes the particles to
move closer to one another with the pores remaining water saturated.
From these considerations it follows that a generally consistent treatment is to relate bentonite water activity to water content, rather than to degree of saturation.
Another consequence of adopting a “matric” view of bentonite (i.e. to include osmotic forces in “matric suction”) is that “matric suction” loses its direct connection with effective stress. This can be illustrated by taking the “osmotic” view: just as the mechanical properties of an aqueous solution (e.g. viscosity) do not depend critically on whether or not it is under (osmotic) pressure, we should not expect e.g. bentonite shear strength to be directly related to swelling pressure.8
Footnotes