It should go without saying that modelers and model developers must justify every feature, mechanism, or component that they use. Failing to do so strongly increases the risk of being fooled by overparameterization rather than gaining insight. The bentonite scientific literature is nonetheless full of incorrect or unjustified model assumptions, several of which have been discussed previously on the blog. Examples include assuming the presence of bulk water, assuming “stack” structures, and assuming that diffusive fluxes from separate domains are additive. Here we discuss yet another unjustified common model component: Stern layers on montmorillonite basal surfaces.
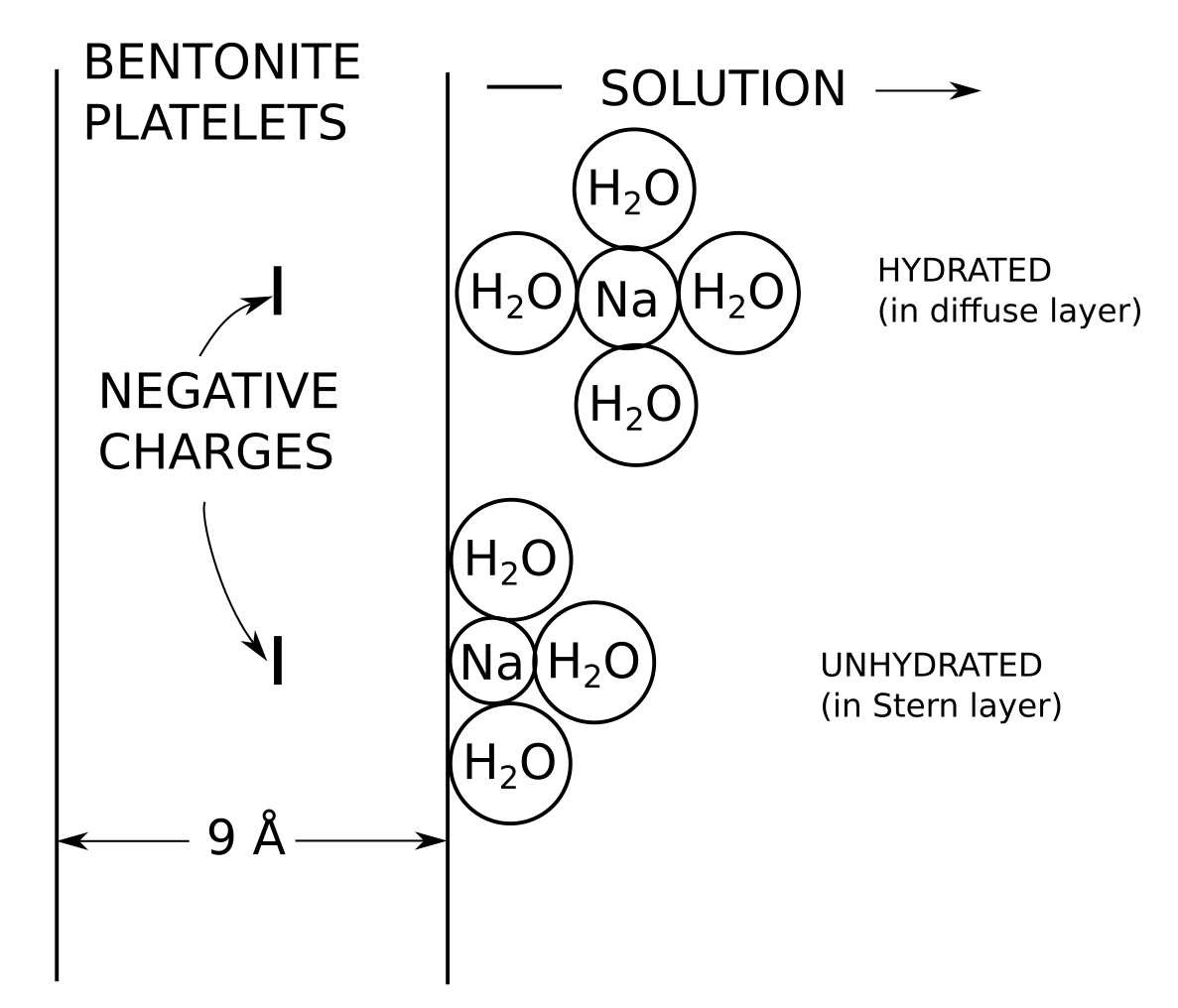
In the bentonite literature, a Stern layer essentially means a layer of “specifically” sorbed ions on the basal surface, as e.g. illustrated here, in a a figure very similar to what is found in Leroy et al. (2006). Illustrations like this are ubiquitous in the literature.

If montmorillonite basal surfaces function roughly as uniform planes of charge we expect the counter-ions to form a diffuse layer, as e.g. described by the Gouy-Chapman model. By introducing a Stern layer, however, many bentonite researchers mean that exchangeable cations in general also interact with basal surfaces by forming immobile surface complexes. Such interactions necessarily involve mechanisms more “chemical” than the pure electrostatic interaction with uniform planes of charge, and the typical description postulates localized “sorption sites”, as illustrated above.
This blog post treats three different main arguments against Stern layers, presented in different sections
- Lack of a coherent description of the surface chemistry
- Deviations from the Gouy-Chapman model have other causes
- Experimental evidence of counter-ion mobility
I want to make clear that this criticism concerns one particular type of surface: the montmorillonite basal surface. Stern layer models are found in many research fields dealing with solid interfaces, and although they have been criticized more generally, here we have no intention of doing so. Likewise, the process of surface complexation is certainly important generally — even in bentonite, e.g. on edge surfaces of montmorillonite particles.1
To better be able to criticize the use of Stern layers on basal surfaces in bentonite modeling, we begin by discussing the origin of the Stern’s model.
Origins of the Stern Layer model
The Stern layer concepts were introduced by Otto Stern2 as an extension of the Gouy-Chapman model. Stern’s main concern was metallic electrodes in electrochemical applications. In such systems, the surface (electrode) potential is externally controlled, and can typically be on the order of 1 volt. It is easily seen that the Gouy-Chapman model predicts nonsense for such surface potentials. For e.g. a 1:1 system, the counter-ion concentration at the surface is enhanced by a factor of the order of \(10^{17}\)(!), as seen directly from the Boltzmann distribution \(c^\mathrm{surf} = c^\mathrm{ext}\cdot e^{e\psi^0/kT}\), where \(\psi^0\) is the surface potential, \(e\) is the elementary charge, \(kT\) the thermal energy, and \(c^\mathrm{ext}\) is the concentration far away from the surface. The main problem is that the Gouy-Chapman model does not account for the finite size of ions, and therefore can accumulate an arbitrary amount of charge at the surface. To remedy this flaw, Stern suggested to divide the interface region into a “compact” layer and a “diffuse” part, with the division located an ionic radius from the electrode surface (sometimes referred to as the outer Helmholtz plane).
In the simplest version of Stern’s model the compact layer is free of charges but act as a plate capacitor with a prescribed capacitance (per unit area) \(K_0\). In the original paper Stern shows that, with electrode potential \(\psi^0 = 1\) V and external 1:1 solution concentration \(c^\mathrm{ext} = 1\) M, such a capacitive layer reduces the potential where the diffuse layer begins to \(\psi^1 = 0.08\) V; lowering the external concentration to \(c^\mathrm{ext} = 0.01\) M gives \(\psi^1 = 0.18\) V. For these calculations, Stern uses a value of \(K_0 = 0.29\;\mathrm{F/m^2}\), adopted from measurements on mercury electrodes. This version of the Stern model is essentially a way to take into account that ions cannot get arbitrarily close to the surface.
Stern also presented more elaborate versions of the model that include adsorption in the compact layer (as a Langmuir adsorption model). It is such mechanisms that is universally referred to as a Stern layer in the bentonite scientific literature. Clearly, such versions are substantially more conceptually complex; rather than to just account for a finite ion size at the first molecular layer, we must now also consider additional chemical interactions that typically are different for different types of ions. We also need to have an idea about the adsorption capacity.
Lack of a coherent description of “specific sorption” on montmorillonite basal surfaces
When using Stern layers for describing montmorillonite basal surfaces, a first thing to note is that the surface potential is not independently controlled for these systems. In contrast to metallic electrodes, montmorillonite is characterized by a fixed surface charge and the problem of accumulating unrealistically large amounts of ions at the interface is significantly mitigated. As pointed by e.g. Norrish and Bolt already in the 1950s, even if we put all counter-ions within the first nanometer adjacent to the surface, the corresponding ion concentration is not larger than approximately 3 M. Here is a illustration of the montmorillonite basal surface on the nm scale, with a representative number of monovalent counter-ions (top layer oxygen atoms are red and the counter-ions blue).3
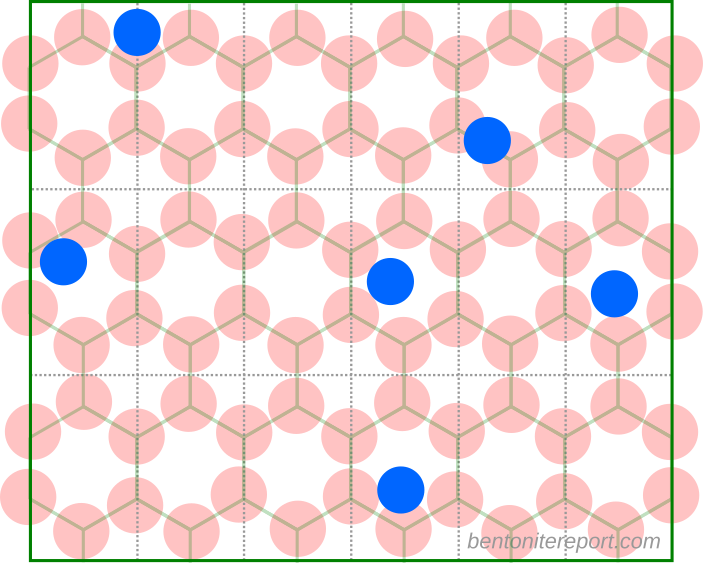
Clearly, there is room to accommodate all ions without running into the problems that was initially addressed by Stern’s model. Of course, solely accounting for the finite size of the ions — as is done in the simplest version of Stern’s model — is always well justified and will in principle improve the description. In particular, a pure diffuse layer model overestimates the capacitance of the surface. As shown in the table below, the introduction of an empty Stern layer “fixes” this problem.
| \(c^\mathrm{ext}\) (M) | \(\psi^1\) (V) | \(c^1\) (M) | \(\psi^0\) (V) | \(K_\mathrm{Stern}\) (\(\mathrm{F}/\mathrm{m ^2}\)) | \(K_\mathrm{DL}\) (\(\mathrm{F}/\mathrm{m ^2}\)) |
| 0.001 | 0.206 | 3.57 | 0.590 | 0.188 | 0.538 |
| 0.01 | 0.148 | 3.59 | 0.532 | 0.209 | 0.748 |
| 0.1 | 0.092 | 3.77 | 0.475 | 0.234 | 1.212 |
| 1 | 0.042 | 5.39 | 0.426 | 0.260 | 2.612 |
Here \(c^\mathrm{ext}\) is the concentration of the 1:1 salt far away from the surface, \(\psi^1\) and \(c^1\) denote the electrostatic potential and the counter-ion concentration, respectively, at the point where the diffuse layer begins (i.e. at the interface to the compact layer), and \(\psi^0\) is the electrostatic potential at the surface. \(K_\mathrm{Stern}\) denotes the corresponding capacitance as calculated from the Stern model, with the choice \(K_0 = 0.29 \;\mathrm{F/m^2}\). \(K_\mathrm{DL}\) is instead the capacitance as calculated from a pure diffuse layer model (in which case the surface potential has the value of \(\psi^1\)). In the calculations are assumed a montmorillonite surface charge of 0.111 \(\mathrm{C}/\mathrm{m ^2}\).
But to simply account for the finite size of the ions by means of an empty compact layer is not how the term Stern layer is used in the bentonite scientific literature. As mentioned, most bentonite researchers mean that parts of the rather sparse collection of ions on the surface interacts chemically (“specific sorption”, “chemisorption”). The question of whether Stern layers on montmorillonite basal surfaces are well motivated thus reduces to what arguments there are for more elaborate chemical mechanisms being active on these surfaces. And descriptions of specific sorption on basal surfaces are really all over the place.
Deshpande and Marshall (1959, 1961) claim that counter-ions are partitioned between (i) chemisorbed ions, which do not contribute to conductivity or activity, (ii) physisorbed ions in a Stern layer, which do not contribute to activity or D.C. conductivity, and (iii) diffuse layer ions, which contribute fully to activity and conductivity. If I interpret their numbers correctly, they state that about 75% — 80% of the counter-ions in pure K-montmorillonite are immobilized. Note that these authors mean that ions in the Stern layer are “physically” adsorbed, while the surface also have “chemically” adsorbed species. Thus, they use the term Stern layer for certain types of physisorption, while stating that ions also bond covalently to the surface.
Shainberg and Kemper (1966, 1967), on the other hand, model ions as either mobile in a diffuse layer or immobile in a Stern layer. They argue that covalently bound ions are “extremely unlikely”, and mean that ions in the Stern layer form “ion pairs” with the surface, as suggested earlier by Heald et al. (1964). They use this idea as a starting point for analyzing differences in exchange selectivity for different monovalent cations in montmorillonite. They claim that “about 20 to 50% of sodium are specifically adsorbed”.
Note that Shainberg and Kemper, just like Deshpande and Marshall, assume Stern layer ions to be “physically” bonded to the surface (i.e. non-covalently), while having a completely different opinion on the presence of chemisorbed ions. Shainberg and Kemper (1966) provide a picture showing the conceptual difference between an ion in the Stern layer (“unhydrated”) and ions in the diffuse layer (“hydrated”) that looks very similar to this
In this context it may be worth to also mention the work of Low and co-workers. Low argued consistently that swelling pressure is not primarily related to the exchangeable ions — something that I strongly disagree with and that I commented briefly on in a previous blog post.4 Directly related to this view, these authors claim that the exchangeable ions for the most part do not dissociate from the surfaces, and in later papers they refer to such ions as being part of a Stern layer.
To me, all the above descriptions seem like little more than speculation. None of these authors discuss how or why e.g. sodium ions (!) are supposed to from ion pairs with a charge center buried far inside the montmorillonite layer, nor how or why they bond covalently with the basal surface. Nevertheless, both Low as well as Shainberg and Kemper seem to have influenced the writings of Sposito, who, in turn, has had quite a huge impact on contemporary descriptions of bentonite. In e.g. Sposito (1992), which specifically discusses montmorillonite (“smectite”), he writes
Despite the long history of continual investigation of the surface and colloid chemistry of smectites (van Olphen, 1977; Sposito, 1984), the structure of the electrical double layer at smectite surfaces and its influence on the rheological properties of smectite suspensions remain topics of lively controversy. One of the most contentious issues is the partitioning of adsorbed monovalent cations among the three possible surface species on the basal planes of smectite particles, such as montmorillonite (see, e.g., Low, 1981, 1987). […] [A] monovalent cation can be adsorbed on the basal planes by three different mechanisms: inner-sphere surface complexes, in which the cation desolvates and is captured by a ditrigonal cavity; outer-sphere surface complexes, in which the cation remains solvated but still is captured by a ditrigonal cavity and immobilized; and the diffuse-ion swarm, in which the cation is attracted to the basal plane, but remains fully dissociated from the smectite surface (Sposito, 1989a, Chap. 7).
The view conveyed here is that exchangeable ions do not interact with montmorillonite basal surfaces as if these, to a first approximation, are planes of uniform charge. Such interaction is only supposed to govern an outer diffuse layer (called a “diffuse-ion swarm” for unclear reasons), and ions are also supposed to interact with the surface by no less than two other “mechanisms”, related to the “ditrigonal cavities”.
Note that while Sposito acknowledges an ongoing “lively controversy” regarding how to describe montmorillonite basal surfaces, he specifies that this debate is limited to how to distribute the exchangeable ions among “three possible surface species.” But, as we will explore below, there is certainly no consensus within colloid chemistry that exchangeable ions are involved in complexation chemistry on the basal surfaces! (I therefore find this way of formulating the “controversy” quite dishonest, to be honest.) For reasons I can’t get my head around, descriptions of “inner-” and “outer-sphere complexes” on montmorillonite basal surfaces are anyway ubiquitous in modern bentonite literature. Let’s therefore take a closer look at how these are introduced.
“Inner-” and “outer-sphere” surface complexes
A description that hardly enlightens me is given in Sposito (1989)5
[The inner- and outer-sphere complexes] constitute the Stern layer on an adsorbent. […] The diffuse-ion swarm and the outer-sphere surface complex mechanisms of adsorption involve almost exclusively electrostatic bonding, whereas inner-sphere complex mechanisms are likely to involve ionic as well as covalent bonding. Because covalent bonding depends significantly on the particular electron configurations of both the surface group and the complexed ion, it is appropriate to consider inner-sphere surface complexation as the molecular basis of the term specific adsorption. Correspondingly, diffuse-ion screening and outer-sphere surface complexation are the molecular basis for the term nonspecific adsorption. Nonspecific refers to the weak dependence on the detailed electron configurations of the surface functional group and adsorbed ion that is to be expected for the interactions of solvated species.
Here, Sposito means that exchangeable ions bond covalently with the montmorillonite basal surface,6 in agreement with Deshpende and Marschall, and in disagreement with Shainberg and Kemper (we have one “extremely unlikely” and one “likely” for covalent bonding…). In contrast to Deshpende and Marschall, however, Sposito means that these “inner-sphere” complexes are part of the Stern layer. To confuse matters even more, Shainberg and Kemper assume their “unhydrated” construct (which corresponds structurally to an “inner-sphere” complex, see above figure) as being part of the Stern layer, but not part of any covalent bonding. Moreover, Shainberg and Kemper assume their “hydrated” construct (corresponding structurally to an “outer-sphere” complex, see above figure) to be part of the diffuse layer, while Sposito wants his “outer-sphere” complexes to be immobile and part of the Stern layer…
Given the above description (and others) it is hard to understand what the difference is supposed to be between an “outer-sphere” complex and an ion in the “diffuse-ion swarm”, other than that the former is simply claimed to be immobilized; both ions are said to interact with “exclusively electrostatic bonding”,7 both are classified as “nonspecific adsorption”, and both are fully hydrated. In my head, this is simply a recipe for achieving an overparameterized model description.
Sposito’s description also makes implicit statements about the montmorillonite basal surface: it contains “surface functional groups” whose specific electron configuration significantly influence covalent bonding, while being insensitive for the formation of “outer-sphere” complexes and the “diffuse ion-swarm”. In Sposito (1984) he suggests that the “functional groups” are groups of oxygen atoms on the surface of the montmorillonite particle (“ditrigonal cavities”) that qualifies as Lewis bases. The presence of atomic substitutions in the octahedral layer is supposed to enhance this Lewis base character
If isomorphic substitution of \(\mathrm{Al}^{3+}\) by \(\mathrm{Fe}^{2+}\) or \(\mathrm{Mg}^{2+}\) occurs in the octahedral sheet, the resulting excess negative charge can distribute itself principally over the 10 surface oxygen atoms of the four silica tetrahedra that are associated through their apexes with a single octahedron in the layer. This distribution of negative charge enhances the Lewis base character of the ditrigonal cavity and makes it possible to form complexes with cations as well as with dipolar molecules.
Note how completely different this whole description is compared to the original Gouy-Chapman conceptual view. Here is implied that montmorillonite basal planes8 cannot be described as a passive layer of charge, but that it is a fully reactive system, including covalent bonding mechanisms.
Frankly, I dismiss the above description of the montmorillonite surface as a Lewis base as pure speculation. I will gladly admit that I am a physicist rather than a chemist, and perhaps I am missing something obvious, but I really don’t see any argumentation behind this description. I am also under the impression that montmorillonite basal planes are relatively chemically stable — that is why they form in the first place, and that is also one reason for why we are interested in using bentonite for e.g. long-term geological waste storage. Furthermore, a meta-argument for dismissing this description is that in later publications we find statements like this, in Sposito (2004):
The \(\mathrm{Na}^+\) that are counterions for the negative structural charge developed as a result of isomorphic substitutions within the clay mineral layer tend to adsorb as solvated species on the basal plane (a plane of hexagonal rings of oxygen ions known as a siloxane surface) near deficits of negative charge originating in the octahedral sheet from substitution of a bivalent cation for \(\mathrm{Al}^{3+}\). This mode of adsorption occurs as a result of the strong solvating characteristics of Na and the physical impediment to direct contact between Na and the site of negative charge posed by the layer structure itself. The way in which this negative charge is distributed on the siloxane surface is not well known, but if the charge tends to be delocalized there, that would also lend itself to outer-sphere surface complexation.
So, 20 years after the surface chemistry of montmorillonite was described as if it was completely understood (Sposito, 1984), the way the negative charge distributes is now described instead as “not well known”… Furthermore, in contrast to earlier statements, the formation of an “outer-sphere complex” is here associated mainly with the hydration properties of sodium. But if the idea of a “surface functional group” is discarded — or at least downplayed — why should a hydrated ion near the surface be described as a surface complex at all?
We note that Sposito (2004) still seem to imply that the “outer-sphere surface complex” is localized and immobile (“adsorbed near deficits of negative charge”) But the evidence is vast that sodium, and several other ions, are quite mobile even in monohydrates (see below).
Deviations from the Gouy-Chapman model do not imply surface complexation
Authors that promote Stern layers on montmorillonite basal surfaces usually rely on the Gouy-Chapman model for describing the diffuse layer part. Lyklema, writing generally on colloid science, explicitly “defends” such an approach
In the following our discussion will be based on the rather pragmatic, though somewhat artificial, subdivision of the solution side of the double layer into two parts: an inner part, or Stern layer where all complications regarding finite ion size, specific adsorption, discrete charges, surface heterogeneity, etc., reside and an outer, Gouy or diffuse layer, that is by definition ideal, i.e. it obeys Poisson-Boltzmann statistics. This model is due to Stern following older ideas of Helmholtz and has over the decades since its inception rendered excellent services, especially in dealing with experimental systems.
Dzombak and Hudson (1995) express a similar attitude
Bolt and co-workers […] investigated in detail the application of the Gouy–Chapman diffuse-layer theory to ion-exchange processes. Their work demonstrated that consideration of electrostatic sorption alone is not sufficient to explain ion-exchange data and that chemisorption (or “specific” sorption) needs to be included in ion-exchange models.
It is not logically consistent to conclude that deviations from the Gouy-Chapman model implies that specific sorption “needs to be included”.9 On the contrary, introducing specific sorption to compensate for a certain model rather than for surface chemical reasons may, in my mind, be a recipe for an overparameterized disaster. I don’t get reassured by statements like this, also from Dzombak and Hudson (1995)
Surface complexation models can be extended to include diffuse-layer sorption. This approach permits their application in modeling the sorption of ions (such as monovalent electrolyte ions) that exhibit weak specific sorption. The generality of such an extended surface complexation approach together with the mathematical power of modern chemical speciation models offers the potential for accurate physicochemical modeling of ion exchange
Reasonably, a complex system may require complex models, but it is certainly dangerous in a modeling context to rely too heavily on “mathematical power” (I guess “numerical power” is the preferred phrase).10
Note that very different attitudes towards the Stern layer concept is found in the colloid science literature, where e.g. Evans and Wennerström (1999) describe it as an “intellectual cul de sac”.11
One way of dealing with these difficulties is to say that the solution layer closest to a charged surface has properties so different from the bulk that it should be treated as a separate entity. This device was introduced in the 1930s by the German electrochemist Stern and the surface layer is commonly referred to as the Stern layer, whose properties are specified by a number of empirical parameters. It is the opinion of the authors of this book that the Stern layer concept is an intellectual cul de sac for the description of electrostatics in colloidal systems. One reason for this point of view is that from modern spectroscopic measurements we know molecular properties are not dramatically changed for a liquid close to a charged surface.
I find it quite perplexing that so many authors in the bentonite scientific community attribute any deviation from the Gouy-Chapman model solely to surface-related mechanisms. The Gouy-Chapman model treats both ions (point particles) and water (a dielectric continuum) in a very simplified manner, and it is clear that “specific ion” effects are ubiquitous, also in systems that lack surfaces. Addressing differences in e.g. selectivity coefficients without considering ion polarizability and hydration, while postulating the existence of localized sorption “sites”, can, to my mind, only lead to incorrect descriptions.
The Poisson-Boltzmann equation is a mean field approximation
Note also that the Poisson-Boltzmann equation — which underlies the Gouy-Chapman model — is only approximate. It is derived by assuming that the electrostatic potential experienced by any ion is the average potential from all other ions (and surfaces). More accurately, the ion distribution around a given ion deviates from the average, as a direct consequence of the presence of the central ion.
Including these ion-ion correlation effects makes the mathematical description considerably more complex. But with the advent of sufficiently powerful computers and algorithms, the electric double layer has been solved basically “exactly”. The “exact” solution may differ strongly from the Poisson-Boltzmann solution, with increasing concentrations towards the surfaces (and consequently a lowering of interlayer midpoint concentrations), and an explicit attractive electrostatic force between the two halves of an interlayer. Using Monte Carlo simulations, Guldbrand et al. (1984) demonstrated that with divalent counter-ions these effects are so large that the system becomes net attractive at a certain interlayer distance, in qualitative disagreement with the Poisson-Boltzmann solution. This effect, which has been thoroughly studied since the 80s, and which we have discussed in several posts on this blog, is the prevailing explanation e.g. for the limited swelling of Ca-montmorillonite.
The lesson here is that observed deviations from predictions of the Poisson-Boltzmann equation not automatically can be taken as evidence for additional active system components, and certainly not as evidence for specific sorption. Note that limited swelling in divalent montmorillonite is explained by the ions being diffusive, not that they are sorbed and immobilized. I cannot overstate the importance of this insight.
It boggles my mind that the entire research area on ion-ion correlations in colloidal systems seems to have made no significant impact on parts of the bentonite scientific community; I seldom find references to works on ion-ion correlation, and when I do it’s quite confused. E.g. Sposito (1992) means that the formation of “quasicrystals”12 is due to “outer-sphere” complexes
The best known example of a montmorillonite quasicrystal is that comprising stacks of four to seven layers. \(\mathrm{Ca}^{2+}\) ions, solvated by six water molecules (outer-sphere surface complex), serve as molecular “cross-links” to help bind the clay layers together through electrostatic forces.
Sure, the ion-ion correlation effect that prevents Ca-montmorillonite from exfoliating is of electrostatic origin, but it is not related to “cross-links” or surface complexes. Sposito furthermore continues by claiming that “even […] Na-montmorillonite” forms “quasicrystals”. Such claim cannot be supported by ion-ion correlation — on the contrary, ion-ion correlation explains why Ca-montmorillonite forms “quasicrystals”, while Na-montmorillonite does not. It is thus relatively clear that Sposito do not refer to ion-ion correlation in the above statement. At the same time, later in the same publication he cites Kjellander et al. (1988) on going beyond the mean-field treatment of the Poisson-Boltzmann equation, and even claims that the Gouy-Chapman model is “completely inaccurate” for systems containing divalent ions. I can only conclude that this is a quite confused description.
Finite-size effects of water molecules
With focus on the first molecular layers at a solid interface, it is clear that finite-size effects of water molecules — which are not treated in the Gouy-Chapman model — reasonably influences the resulting ion distribution. This influence is manifested both as a steric effect — there can only be a discrete number of water molecules between the ion and the surface — and as an effect of how strongly a certain ion is hydrated.
Water molecules are treated explicitly e.g. in molecular dynamics (MD) simulations of montmorillonite/water interfaces, and here are results from simulating a three water-layer interlayer of Na-montmorillonite, from Hedström and Karnland (2012)13

Sodium is seen to accumulate “between” the water layers; in the above illustration we have also included schematic illustrations of the molecular configurations, as conceived by Shainberg and Kemper (1966) (shown earlier). As stated earlier, Shainberg and Kemper (1966) refer to these as “hydrated” and “unhydrated”, but they are clearly the same type of configurations that e.g. Sposito (1992) and Dzombak and Hudson (1995) call “outer-” and “inner-sphere” complexes.
While the above mentioned authors mean that these “complexes” involve specific interactions between ions and surface, the MD simulation suggests that such structures are mainly a consequence of the finite-size of the molecules and ions. In particular, the MD results do not support the idea that these structures depend critically on a specific, non-electrostatic, ion–surface interaction. Indeed, the simulations explicitly treat also the atoms of the montmorillonite layer, which could make it difficult to judge whether the appearance of “complexes” mainly is related to water–ion or ion–surface interactions. But note that Hedström and Karnland (2012) simulate two different systems: one where the montmorillonite charge is put on specific atoms in the octahedral sheet (Mg for Al substitutions), and one where it is distributed on all Al atoms (as a fraction of the elementary charge). Both systems have essentially an identical atomic configuration in the interlayer, which strongly suggest that no critical ion–surface interaction is involved in forming “outer-” and “inner-sphere complexes” (i.e. they really are not surface complexes). I am not aware of any published simulation where the basal surface is represented as a uniform sheet of charge while water molecules are treated explicitly, but I am convinced that “outer-” and “inner-sphere complexes” would appear also in such a simulation.
Regarding MD simulations of montmorillonite interlayers, you can also simply observe them to convince yourself that the counter-ions are not in any reasonable sense immobilized. These types of simulations are routinely used to calculate (quite significant) interlayer diffusion coefficients, for crying out loud!
Experimental evidence of counter-ion mobility
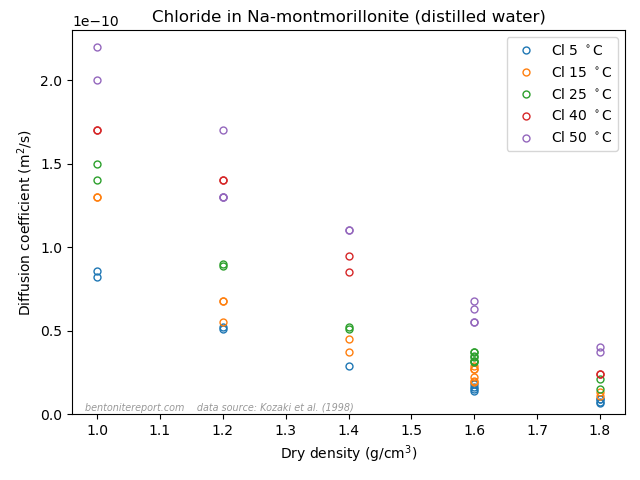
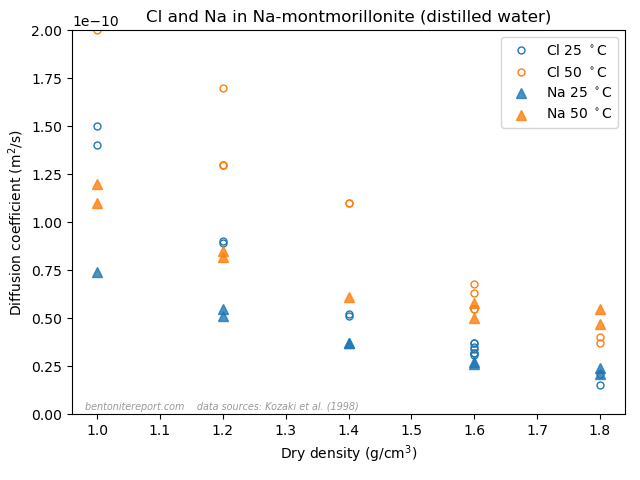
A final argument for why Stern layers on montmorillonite basal surfaces are unjustified is the vast amount of empirical evidence of counter-ion mobility. We have discussed several diffusion studies in earlier blog posts that show that many ions (Na, Cl, K, Sr, I, Cs, Ca,…) have a significant mobility even in very dense systems, dominated by bi- or monohydrated interlayers. In the previous post, we brought up the following result
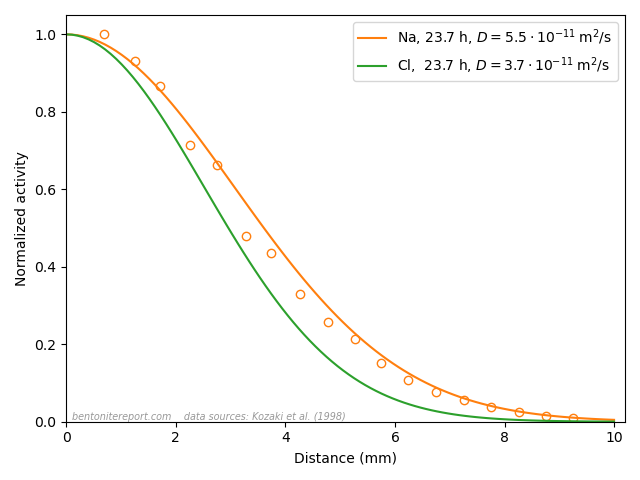
This figure shows the resulting concentration profiles in two diffusion experiments where sodium and chloride tracers, respectively, have diffused from an initial planar source for the same amount of time (23.7 h), in samples of pure Na-montmorillonite of dry density 1.8 \(\mathrm{g/cm^3}\), equilibrated with deionized water. This result was used previously to dismiss the ludicrous idea that these two ions are supposed to migrate in separate parts of the pore volume, exposed to completely different mechanisms. In the same vein, this result can be used to dismiss the idea of a Stern layer on basal surfaces.
Sodium, which is universally acknowledged to reside in the interlayers, is here demonstrated to diffuse just fine in bi- and monohydrated interlayers. As chloride, which also resides in the interlayers (despite all talk of “anion-accessible porosity”), behave essentially identical, it is quite far-fetched to assume any significant surface complexation mechanism. And anyone who argues for that these tracers actually do not diffuse in the interlayers should be reminded of the seeming “uphill” diffusion experiment,14 which is performed at even higher density, and where the “uphill” diffusion direction once and for all proves that the transport occurs in interlayers.
Strangely, many authors nowadays seem to promote both Stern layers and interlayer mobility in bentonite. Various simulation codes has been modified for this possibility, and there are several examples of researchers pointing out a possibility of “Stern layer diffusion”. I think these authors should carefully examine their chain of assumptions: Surface complexation in a Stern layer (i.e. sorption) is initially suggested to explain e.g. why breakthrough times in cation through-diffusion tests are relatively long as compared with the steady-state flux (i.e. why “\(D_e\)” can be considerably larger than”\(D_a\)”). With evidence for that the “sorbed” ions actually dominate the mass transfer, the sorption mechanism is not reconsidered, but yet another mechanism is suggested: Stern layer mobility… Reasonably, such an approach is not adequate for developing models; researchers employing it should critically consider the intended purpose of a Stern layer component.
Counter-ion mobility is also related to swelling pressure. Bentonite swelling pressure is difficult to describe generally, and I have written a whole series of blog posts on the subject, but it is clear that measured swelling pressures in e.g. moderately dense Na- and Li-montmorillonites is quite well described by the Poisson-Boltzmann equation. As this set of conditions (not too dense clay, simple monovalent ions) are exactly those for which we expect the Poisson-Boltzmann equation to be adequate, this is a strong indication that all counter-ions contribute to the pressure.15 Also, the limited swelling in e.g. Ca-montmorillonite, as previously discussed, is explained by ion-ion correlation effects where all ions are included in the diffuse layer.
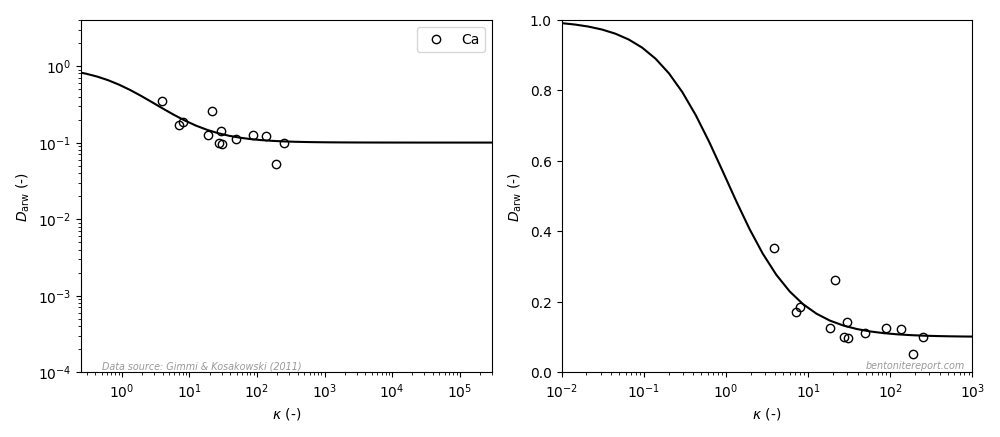
Finally, we can take a look at salt exclusion from compacted bentonite. The magnitude of salt exclusion is directly related to the amount of mobile counter-ions. Thus, if most of the counter-ions were immobilized in a Stern layer, bentonite should show small exclusion effects. In contrast, the empirical results for e.g. chloride exclusion in sodium dominated bentonite indicate, again, that all counter-ions are part of a diffuse layer.

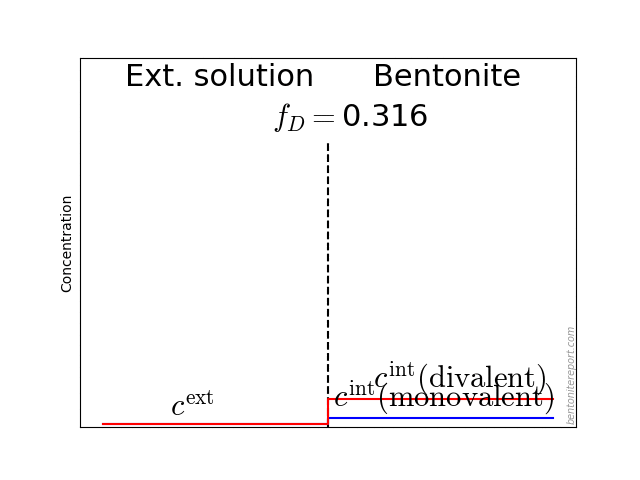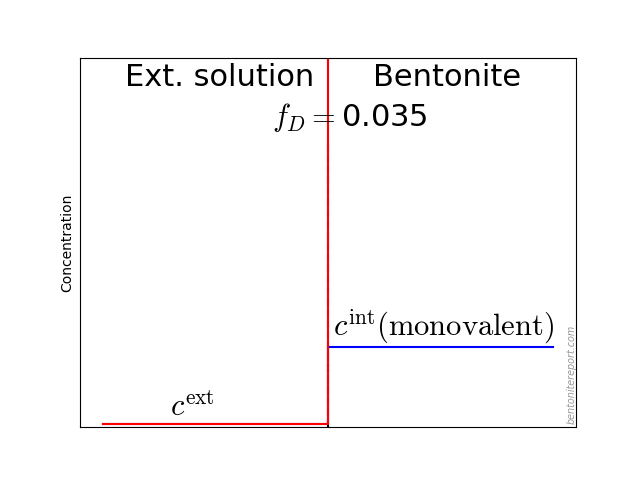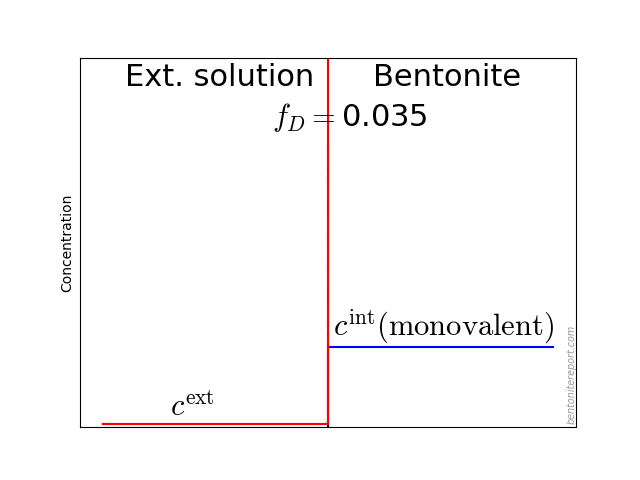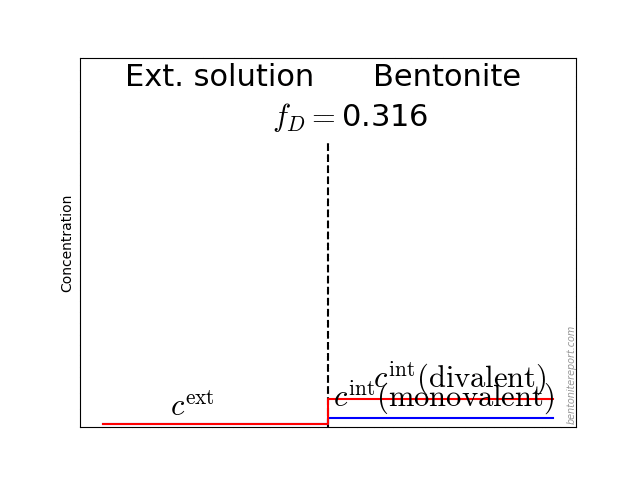
This diagram shows the relative amount of chloride in the bentonite as a function of \(c^\mathrm{ext}/c_\mathrm{IL}\), where \(c^\mathrm{ext}\) is the external salt concentration and \(c_\mathrm{IL}\) is the amount exchangeable cations, expressed as a monovalent interlayer concentration. The experimental data is from Van Loon et al. (2007), which we reevaluated and examined in detail in a previous blog post. The lines are the result from applying the “ideal” Donnan formula with various amounts of the counter-ions assumed diffusive. For details on Donnan theory, see this blog post.
Although the experimental data show considerable scatter, there is nothing in this plot that suggests that a fraction of the counter-ions are immobilized. And the quality of this data is certainly good enough to directly dismiss models that assume that the major part of ions are immobilized in a Stern layer.
Footnotes
[1] I find it quite frustrating that many descriptions in the literature only refer abstractly to “mineral surfaces” rather than specifically addressing montmorillonite. At the same time it is often clear from the context that statements regarding “mineral surfaces” should be understood as applicable to montmorillonite basal surfaces. I would much appreciate if researchers promoting Stern layers on basal surfaces would provide descriptions for specific systems, e.g. pure Na-Ca-montmorillonite.
[2] Otto Stern is a fascinating character in the history of science, most famous for the Stern-Gerlasch experiment, that helped pave the way for quantum mechanics. I highly recommend this lecture by the late Sandip Pakvasa. An example of its contents:
A note on Stern’s style of working: He always had a cigar in one hand, and he left actual work with hands to others, as he did not trust his own manual dexterity! […] He described the beneficial effects of a large wooden hammer that he kept in his lab and used it to threaten the apparatus if it did not behave! (apparently it worked!)
[3] Note that di-valent counter-ions would be even more sparsely distributed than this.
[4] It may be worth discussing my objections against the work of Low and co-workers in more detail in a future blog post.
[5] This is a general discussion on sorption on mineral surfaces, and is cited from the second edition of the book (2008). There is also a “Thired” edition.
[6] This particular description is general (for “adsorbents”), but since Sposito, as well as a large part of the contemporary bentonite scientific community, claim that “inner-sphere” complexes are present on montmorillonite basal surfaces, we can conclude that they mean that covalent bonding occur on such surfaces.
[7] Sure, the full quotation is “almost exclusively electrostatic bonding”, but what is a reader supposed to do with that? Such vague and sloppy scientific writing annoys me.
[8] Again, the discussion is on general “mineral surfaces”, but from other writings it is clear that this is supposed to apply to the montmorillonite basal surface.
[9] I furthermore don’t believe that “Bolt and co-workers” concluded that specific sorption “needs” to be included, but that this rather is an interpretation made by Dzombak and Hudson themselves. Bolt considered and downplayed the Stern Layer already in the mid 50s, and although he indeed has expressed positive attitudes (seriously, these guys just write too much!), he continued to downplay its significance in e.g. Bolt (1979), writing “In conclusion it appears justified to assume that for homoionic clays saturated with common ions, if hydrated, the Stern layer will be an “empty” Stern layer according to the terminology of Grahame (1947).”
[10] Note also that the perspective in this quotation is that specific sorption models can be complemented with diffuse layer features — i.e. the existence of sorption “sites” is assumed a priori. But Dzombak and Hudson (1995) never really discuss the nature of such “sites” on montmorillonite basal surfaces, but relies on Sposito’s speculations about “inner-” and “outer-surface” complexes.
[11] Note that Stern’s original paper actually is from 1924. I also suspect that Stern would object to being labeled an “electrochemist”.
[12] The terminology here is quite messy, and other authors may use other terms such as “tactoids”, see this post for a further discussion.
[13] This study was thoroughly discussed in a blog post on MD simulations and anion exclusion.
[14] Anyone making this argument should also provide a plausible suggestion for where a significant non-interlayer pore structure is located at these extreme densities.
[15] The pressure in these types of calculations can be related to the interlayer midpoint concentration. But this does not mean that not all counter-ions are involved in the process.