Mu07 actually report 9 more data points, but these originate from Muurinen et al. (2004) (which we have already assessed). This is not fully acknowledged in Mu07, but below I try to sort out the status of all data presented. We refer to Muurinen et al. (2004) as Mu04.
In similarity to Mu04, Mu07 is an equilibrium study (i.e. not a diffusion study) performed on purified “MX-80” bentonite. One of the main objectives in Mu07 is to investigate possible influence of sample preparation on the chloride equilibrium concentrations. The samples in Mu07 cover a large density range (0.6 — 1.5 g/cm3), but were all equilibrated with a single type of solution: 0.1 M NaCl.
Originally conducted and already reported tests
Mu07 state that the study is a continuation of the investigations
presented in Mu04 and present data on five different sets of samples,
prepared and equilibrated using different methods (labeled A —
E). What is not explicitly stated — but what is obvious if comparing
tables 1 and 2 in Mu04 with table 1 in Mu07 — is that sample sets D
and E are the same as previously reported in Mu04.
I find this quite remarkable, since two of these samples were
dismissed as “not reliable” in Mu04
(In my
assessment, I dismissed all tests in Mu04); here the same results
— which show an increase in equilibrium chloride content with
density — are not only re-reported, but modeled! The authors don’t
even seem aware that they have previously discarded the samples,
writing: “Surprisingly, it seems that the concentrations in the
sample types D and E start to increase at the highest
densities“. Furthermore, one of the (previously reported) data points
of sample set E, which have a clay concentration larger than
the corresponding concentration of the equilibrating solution, is
not included in Mu07. Needless to say, excluding data points
without motivation, or including previously discarded data is not good
scientific practice.
As the sample overview table in Mu07 also has some misprints,1 I here present a (hopefully) correct version that also indicates original publication (for the indicated sample-IDs, see the assessment of Mu04).
Type
Density (g/cm3)
Time d.w. (days)
Time 0.1 M (days)
Clay conc. (mM)
origin/remark
A
0.625
18+35
217
37
Mu07
A
0.812
18+35
217
29
Mu07
A
1.200
18+35
217
20
Mu07
B
0.670
222
107
35
Mu07
B
0.937
222
107
23
Mu07
B
1.389
222
107
15
Mu07
C
0.622
30
36
48
Mu07
C
0.731
30
36
30
Mu07
C
1.113
30
36
17
Mu07
C
1.382
30
36
14
Mu07
C
1.517
30
36
12
Mu07
D
0.754
0
40
65
Mu04 (S2-02)
D
0.855
0
40
39
Mu04 (S2-21)
D
1.273
0
36
22
Mu04 (S2-04)
D
1.636
0
36
24
Mu04 (S2-17; deemed “not reliable” in Mu04)
D
1.764
0
85
48
Mu04 (S2-18; deemed “not reliable” in Mu04)
E
0.750
0
12(?)
109
Mu04 (not included!)
E
0.875
0
12
61
Mu04
E
1.225
0
12
25
Mu04
E
1.516
0
12
12
Mu04
E
1.543
0
12
14
Mu04
In the following, the focus is solely on samples sets A — C (as
mentioned, the others have already been
assessed).
Material
The material appear to be the same as used in Mu04. I therefore refer to the assessment of that study for a detailed discussion. In brief, the material is purified “MX-80” bentonite, with a montmorillonite content above 90% and about 90% sodium as exchangeable cation.
Samples
Samples in the three different sample sets A — C were prepared in different ways. For set A, the clay was initially dispersed in deionized water at quite low density. After an equilibration time of 18 days (which included ultrasound treatment), the dispersion was slowly squeezed to achieve the intended densities. This squeezing phase lasted 35 days, after which the samples were contacted with 0.1 M NaCl and equilibrated for 217 days.
Samples in set B were prepared in the same type of sample holder as
those in set A, but the bentonite powder was directly compacted to the
desired density, and the samples were water saturated by contact with
deionized water for 222 days (!). Thereafter, the samples were
contacted with 0.1 M NaCl and equilibrated for 107 days.
The external solution was not circulated in the preparation of samples
in sets A and B. In contrast, samples in set C were prepared in cells
with external circulation. The bentonite powder was directly
compacted to the desired density, and the samples were water
saturated by contact with (circulating) deionized water for 30
days. The samples were then equilibrated with (circulating) 0.1 M NaCl
solution for 36 days.
Even if the preparation protocols are described quite detailed in Mu07, we are not given any information on sample geometry. We are not even told if the samples have the same geometry! (Given that they were prepared in different types of equipment, different geometries may certainly be the case.) Without knowledge on e.g. the characteristic diffusion lengths, it is impossible to assess e.g. whether the adopted equilibration times are adequate. Reasonably, the size of the samples are on the cm scale, and since the equilibration times are very long, we can guess that they have had time to equilibrate. This is in contrast to the samples in Mu04, which we have reason to suspect have not been completely equilibrated, as discussed in the assessment of that study. (Note that these samples are included in Mu07, as sample sets D and E.)
Mu07 does not provide any information on how sample density was
measured. Since we neither know the dimensions of the samples it is
therefore impossible to estimate any uncertainty of the reported
densities.
Chloride equilibrium concentrations
The following plot summarizes the reported chloride equilibrium
concentrations and corresponding densities in sample sets A — C.
Although the data show some significant scatter (e.g. for the two
lowest densities in sample set C), the main impression is that the
three different ways of preparing and equilibrating samples result in
quite similar values for the chloride equilibrium concentrations. Thus,
even if we know little about the samples, this coherence in the
results indicates that they have been properly equilibrated.
Possible interface excess salt
As we have discussed in
severalpreviousblog
posts, when performing equilibrium tests it is important to handle
the possibility that the samples have an increased salt content in the
interface regions. In the assessment of Mu04, my guess was that the
samples had not been handled specifically to deal with this possible
measuring artifact, and I neither see any reason to believe that this
issue has been addressed in sample sets A — C (we can, however, rule
out that too much salt entered these samples during saturation, since
deionized water was used in this phase).
The possible influence of interface excess depends, apart from general
sample treatment, on e.g. sample thickness and the concentration of
the equilibrating external solution. As noted above, we have no
information on sample thickness, but the external concentration is in
this regard quite low (we showed in an earlier post that the problem
of interface excess salt becomes more severe for thin samples and low
external concentrations). Therefore, we can certainly not exclude the
possibility that the reported equilibrium concentrations are
systematically overestimated due to possible influence of an interface
excess, especially for the denser samples (see
here for details on this).
An argument against that interface excess has significantly influenced
the results is the similar result for the three different sample
sets. Of course, this depends on how similar (or dissimilar) the
samples in the different sets are, of which we have no
information. Under any circumstance, it is very clear that Mu07
provides too little information to fully rely on the reported values.
Summary and verdict
From one perspective, Mu07 is a very straightforward study: samples of
purified bentonite (almost pure Na-montmorillonite) at various density
have been equilibrated with a single type of external solution (0.1 M
NaCl). The results also look reasonably coherent. However, the paper
contains way too little information on e.g. sample geometry and how
density and concentration were measured to fully rely on the
results. In particular, we cannot rule out a systematic overestimation
due to influence of interface excess salt. Furthermore, the main
reason to believe that equilibrium was achieved, is the
similarity between the different test sets.
My decision, however, is to keep these result to use e.g. for possible
qualitative process understanding (specifically, chloride exclusion).
But I will certainly keep in mind the quite extensive lack of
information associated with this data.
Vl07 is centered around a set of through-diffusion tests in “KWK” bentonite samples of nominal dry densities 1.3 g/cm3, 1.6 g/cm3, and 1.9 g/cm3. For each density, chloride tracer diffusion tests were conducted with NaCl background concentrations 0.01 M, 0.05 M, 0.1 M, 0.4 M, and 1.0 M. In total, 15 samples were tested. The samples are cylindrical with diameter 2.54 cm and height 1 cm, giving an approximate volume of 5 cm3. We refer to a specific test or sample using the nomenclature “nominal density/external concentration”, e.g. the sample of density 1.6 g/cm3 contacted with 0.1 M is labeled “1.6/0.1”.
After maintaining steady-state, the external solutions were replaced
with tracer-free solutions (with the same background concentration),
and tracers in the samples were allowed to diffuse out. In this way,
the total tracer amount in the samples at steady-state was
estimated. For tests with background concentrations 0.01 M, 0.1 M, and
1.0 M, the outflux was monitored in some detail, giving more
information on the diffusion process. After finalizing the tests, the
samples were sectioned and analyzed for stable (non-tracer)
chloride. In summary, the tests were performed in the following
sequence
Saturation stage
Through-diffusion stage
Transient phase
Steady-state phase
Out-diffusion stage
Sectioning
Uncertainty of samples
The used bentonite material is referred to as “Volclay KWK”. Similar
to
“MX-80”, “KWK” is just a brand name (it seems to be used mainly
in wine and juice
production). In contrast to “MX-80”, “KWK” has been used in
only a fewresearchstudies related to
radioactive waste storage. Of the studies I’m aware, only
Vejsada et
al. (2006) provide some information relevant
here.1
Vl07 state that “KWK” is similar to “MX-80” and present a table with chemical composition and exchangeable cation population of the bulk material. As the chemical composition in this table is identical to what is found in various “technical data sheets”, we conclude that it does not refer to independent measurements on the actual material used (but no references are provided). I have not been able to track down an exact origin of the stated exchangeable cation population, but the article gives no indication that these are original measurements (and gives no reference). I have found a specification of “Volclay bentonite” in this report from 1978(!) that states similar numbers (this document also confirms that “MX-80” and “KWK” are supposed to be the same type of material, the main difference being grain size distribution). We assume that exchangeable cations have not been determined explicitly for the material used in Vl07.
In a second table, Vl07 present a mineral composition of “KWK”, which I assume has been determined as part of the study. But this is not fully clear, as the only comment in the text is that the composition was “determined by XRD-analysis”. The impression I get from the short material description in Vl07 is that they rely on that the material is basically the same as “MX-80” (whatever that is).
Montmorillonite content
Vl07 state a smectite content of about
70%. Vejsada et
al. (2006), on the other hand, state a smectite content of 90%,
which is also stated in the 1978 specification of “Volclay
bentonite”. Note that 70% is lower and 90% is higher than any
reported montmorillonite content in “MX-80”. Regardless whether or
not Vl07 themselves determined the mineral content, I’d say that the
lack of information here must be considered when estimating an
uncertainty on the amount of montmorillonite (“smectite”) in the
used material. If we also consider the claim that “KWK” is similar
to “MX-80”, which has a documented montmorillonite content
in the range 75 — 85%, an uncertainty range for “KWK” of 70 —
90% is perhaps “reasonable”.
Cation population
Vl07 state that the amount exchangeable sodium is in the range 0.60 —
0.65 eq/kg, calcium is in the range 0.1 — 0.3 eq/kg, and magnesium is
in the range 0.05 — 0.2 eq/kg. They also state a cation exchange
capacity in the range 0.76 — 1.2 eq/kg, which seems to have been
obtained from just summing the lower and upper limits, respectively,
for each individual cation. If the material is supposed to be similar
to “MX-80”, however, it should have a cation exchange capacity in
the
lower regions of this range. Also,
Vejsada et
al. (2006) state a cation exchange capacity of 0.81 eq/kg. We
therefore assume a cation exchange capacity in the range 0.76 — 0.81,
with at least 20% exchangeable divalent ions.
Soluble accessory minerals
According to Vl07, “KWK” contains substantial amounts of accessory carbonate minerals (mainly calcite), and Vejsada et al. (2006) also state that the material contains calcite. The large spread in calcium and magnesium content reported for exchangeable cations can furthermore be interpreted as an artifact due to dissolving calcium- and magnesium minerals during the measurement of exchangeable cations (but we have no information on this measurement). Vl07 and Vejsada et al. (2006) do not state any presence of gypsum, which otherwise is well documented in “MX-80”. I do not take this as evidence for “KWK” being gypsum free, but rather as an indication of the uncertainty of the composition (the 1978 specification mentions gypsum).
Sample density
Vl07 don’t report measured sample densities (the samples are ultimately sectioned into small pieces), but estimate density from the water uptake in the saturation stage. The reported average porosity intervals are 0.504 — 0.544 for the 1.3 g/cm3 samples, 0.380 — 0.426 for the 1.6 g/cm3 samples, and 0.281 — 0.321 for the 1.9 g/cm3 samples. Combining these values with the estimated interval for montmorillonite content, we can derive an interval for the effective montmorillonite dry density by combining extreme values. The result is (assuming grain density 2.8 g/cm3, adopted in Vl07).
Sample density (g/cm3)
EMDD interval (g/cm3)
1.3
1.04 — 1.32
1.6
1.36 — 1.67
1.9
1.67 — 1.95
These intervals must not be taken as quantitative estimates, but as giving an idea of the uncertainty.
Uncertainty of external solutions
Samples were water saturated by first contacting them from one side with the appropriate background solution (NaCl). From the picture in the article, we assume that this solution volume is 200 ml. After about one month, the samples were contacted with a second NaCl solution of the same concentration, and the saturation stage was continued for another month. The volume of this second solution is harder to guess: the figure shows a smaller container, while the text in the figure says “200 ml”. The figure shows the set-up during the through-diffusion stage, and it may be that the containers used in the saturation stage not at all correspond to this picture. Anyway, to make some sort of analysis we will assume the two cases that samples were contacted with solutions of either volume 200 ml, or 400 ml (200 ml + 200 ml) during saturation.
The through-diffusion tests were started by replacing the two saturating solutions: on the left side (the source) was placed a new 200 ml NaCl solution, this time spiked with an appropriate amount of 36Cl tracers, and on the right side (the target) was placed a fresh, tracer free NaCl solution of volume 20 ml. The through-diffusion tests appear to have been conducted for about 55 days. During this time, the target solution was frequently replaced in order to keep it at a low tracer concentration. The source solution was not replaced during the through-diffusion test.
As (initially) pure NaCl solutions are contacted with bentonite that contains significant amounts of calcium and magnesium, ion exchange processes are inevitably initiated. Thus, in similarity with some of the earlierassessed studies, we don’t have full information on the cation population during the diffusion stages. As before, we can simulate the process to get an idea of this ion population. In the simulation we assume a bentonite containing only sodium and calcium, with an initial equivalent fraction of calcium of 0.25 (i.e. sodium fraction 0.75). We assume sample volume 5 cm3, cation exchange capacity 0.785 eq/kg, and Ca/Na selectivity coefficient 5.
Below is shown the result of equilibrating an external
solution of either 200 or 400 ml with a sample of density 1.6 cm3/g,
and the corresponding result for density 1.3 cm3/g and external volume
400 ml. As a final case is also displayed the result of first
equilibrating the sample with a 400 ml solution, and then replacing it
with a fresh 200 ml solution (as is the procedure when the
through-diffusion test is started).
Although the results show some spread, these simulations make it relatively clear that the ion population in tests with the lowest background concentration (0.01 M) probably has not changed much from the initial state. In tests with the highest background concentration (1.0 M), on the other hand, significant exchange is expected, and the material is consequently transformed to a more pure sodium bentonite. In fact, the simulations suggest that the mono/divalent cation ratio is significantly different in all tests with different background concentrations.
Note that the simulations do not consider possible dissolution of accessory minerals and therefore may underestimate the amount divalent ions still left in the samples. We saw, for example, that the material used in Muurinen et al. (2004) still contained some calcium and magnesium although efforts were made to convert it to pure sodium form. Note also that the present analysis implies that the mono/divalent cation ratio probably varies somewhat in each individual sample during the course of the diffusion tests.
Direct measurement of clay concentrations
Chloride
clay concentration profiles were measured in all samples after
finishing the diffusion tests, by dispersing sample sections in
deionized water. Unfortunately, Vl07 only present this chloride
inventory in terms of “effective” or
“Cl-accessible porosity”, a concept often encountered in
evaluation of diffusivity. However, “effective porosity” is
not what is measured, but is rather an interpretation of
the evaluated amount of chloride in terms of a certain pore volume
fraction. Vl07 explicitly define effective porosity as
\(V_\mathrm{Cl}/V_\mathrm{1g}\), where \(V_\mathrm{1g}\) is the “volume
of a unit mass of wet bentonite”, and \(V_\mathrm{Cl}\) is the “volume
of the Cl-accessible pores of a unit mass of bentonite”. While
\(V_\mathrm{1g}\) is accessible experimentally, \(V_\mathrm{Cl}\) is
not. Vl07 further “derive” a formula for the effective porosity
(called \(\epsilon_\mathrm{eff}\) hereafter)
where \(n’_\mathrm{Cl}\) is the amount chloride per mass bentonite, \(\rho_\mathrm{Rf}\) is the density of the “wet” bentonite, and \(C_\mathrm{bkg}\) is the background NaCl concentration.2 In contrast to \(V_\mathrm{Cl},\) these three quantities are all accessible experimentally, and the concentration \(n’_\mathrm{Cl}\) is what has actually been measured. For a result independent of how chloride is assumed distributed within the bentonite, we thus multiply the reported values of \(\epsilon_\mathrm{eff}\) by \(C_\mathrm{bkg}\), which basically gives the (experimentally accessible) clay concentration
Here we also have divided by sample porosity, \(\phi\), to relate the clay concentration to water volume rather than total sample volume. Note that eq. 2 is not derived from more fundamental quantities, but allows for “de-deriving” a quantity more directly related to measurements. (I.e., what is reported as an accessible volume is actually a measure of the clay concentration.)
It is, however, impossible (as far as I see) to back-calculate the actual value of \(n’_ \mathrm{Cl}\) from provided formulas and values of \(\epsilon_\mathrm{eff}\), because masses and volumes of the sample sections are not provided. Therefore, we cannot independently assess the procedure used to evaluate \(\epsilon_\mathrm{eff}\), and simply have to assume that it is adequate.3 Here are the reported values of \(\epsilon_\mathrm{eff}\) for each test, and the corresponding evaluation of \(\bar{C}\) using eq. 2 (column 3)
*) The table in Vl07 says 0.076, but the concentration profile diagram says 0.090. **) The table in Vl07 says 0.16, but this must be a typo.
When using eq. 2 we have adopted porosities 0.536, 0.429, and 0.322,
respectively, for densities 1.3 g/cm3, 1.6 g/cm3, and 1.9 g/cm3.
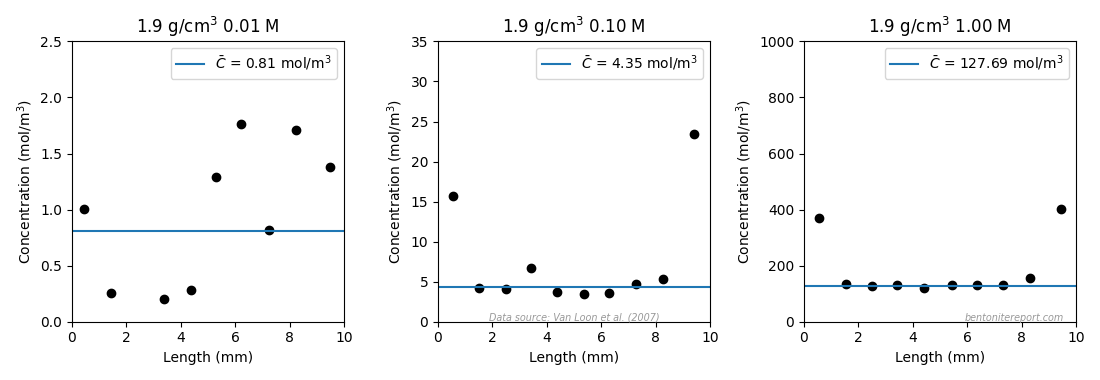
The tabulated \(\epsilon_\mathrm{eff}\) values are evaluated as averages of the clay concentration profiles (presented as effective porosity profiles), which look like this for the samples exposed to background concentrations 0.01 M, 0.1 M and 1.0 M (profiles for 0.05 M and 0.4 M are not presented in Vl07)
The chloride concentration increases near the interfaces in all samples; we have discussed this interface excess effect in previousposts. Vl07 deal with this issue by evaluating the averages only for the inner parts of the samples. I performed a similar evaluation, also presented in the above figures (blue lines). In this evaluation I adopted the criterion to exclude all points situated less than 2 mm from the interfaces (Vl07 seem to have chosen points a bit differently). The clay concentration reevaluated in this way is also listed in the above table (last column). Given that I have only used nominal density for each sample (I don’t have information on the actual density of the sample sections), I’d say that the re-evaluated values agree well with those de-derived from reported \(\epsilon_\mathrm{eff}\). One exception is the sample 1.9/0.01, which is seen to have concentration points all over the place (or maybe detection limit is reached?). While Vl07 choose the lowest three points in their evaluation, here we choose to discard this result altogether. I mean that it is rather clear that this concentration profile cannot be considered to represent equilibrium.
As the reevaluation gives similar values as those reported, and since
we lack information for a full analysis, we will use the values
de-derived from reported \(\epsilon_\mathrm{eff}\) in the continued
assessment (except for sample 1.9/0.01).
Diffusion related estimations
Vl07 determine diffusion parameters by fitting various mathematical expressions to flux data.4 Parameters fitted in this way generally depend on the underlying adopted model, and we have discussed how equilibrium concentrations can be extracted from such parameters in an earlier blog post. In Vl07 it is clear that the adopted mathematical and conceptual model is the effective porosity diffusion model. When first presented in the article, however, it is done so in terms of a sorption distribution coefficient (\(R_d\)) that is claimed to take on negative values for anions. The presented mathematical expressions therefore contain a so-called rock capacity factor, \(\alpha\), which relates to \(R_d\) as \(\alpha = \phi + \rho_d\cdot R_d\). But such use of a rock capacity factor is a mix-up of incompatible models that I have criticized earlier. However, in Vl07 the description involving a sorption coefficient is in words only — \(R_d\) is never brought up again — and all results are reported, interpreted and discussed in terms of effective (or “chloride-accessible”) porosity, labeled \(\epsilon\) or \(\epsilon_\mathrm{Cl}\). We here exclusively use the label \(\epsilon_\mathrm{eff}\) when referring to formulas in Vl07. The mathematics is of course the same regardless if we call the parameter \(\alpha\), \(\epsilon\), \(\epsilon_\mathrm{Cl}\), or \(\epsilon_\mathrm{eff}\).
Mass balance in the out-diffusion stage
Vl07 measured the amount of tracers accumulated in the two reservoirs during the out-diffusion stage. The flux into the left side reservoir, which served as source reservoir during the preceding through-diffusion stage, was completely obscured by significant amounts of tracers present in the confining filter, and will not be considered further (also Vl07 abandon this flux in their analysis). But the total amount of tracers accumulated in the right side reservoir, \(N_\mathrm{right}\),5 can be used to directly estimate the chloride equilibrium concentration.
The initial concentration profile in the out-diffusion stage is linear (it is the steady-state profile), and the total amount of tracers, \(N_\mathrm{tot}\),6 can be expressed
where \(\bar{c}_0\) is the initial clay concentration at the left side interface, and \(V_\mathrm{sample}\) (\(\approx\) 5 cm3) is the sample volume.
A neat feature of the out-diffusion process is that two thirds of the
tracers end up in the left side reservoir, and one third in the right
side reservoir, as illustrated in this simulation
\(\bar{c}_0\) can thus be estimated by using
\(N_\mathrm{tot} = 3\cdot N_\mathrm{right}\) in eq. 3, giving
where \(c_\mathrm{source}\) is the tracer concentration in the left side reservoir in the through-diffusion stage.7 Although eq. 4 depends on a particular solution to the diffusion equation, it is independent of diffusivity (the diffusivity in the above simulation is \(1\cdot 10^{-10}\) m2/s). Eq. 4 can in this sense be said to be a direct estimation of \(\bar{c}_0\) (from measured \(N_\mathrm{right}\)), although maybe not as “direct” as the measurement of stable chloride, discussed previously.
Vl07 state eq. 4 in terms of a “Cl-accessible porosity”, but this is still just an interpretation of the clay concentration; \(\bar{c}_0\) is, in contrast to \(\epsilon_\mathrm{eff}\), directly accessible experimentally in principle. From the reported values of \(\epsilon_\mathrm{eff}\) we may back-calculate \(\bar{c}_0\), using the relation \(\bar{c}_0 / c_\mathrm{source} = \epsilon_\mathrm{eff}/\phi\). Alternatively, we may use eq. 4 directly to evaluate \(\bar{c}_0\) from the reported values of \(N_\mathrm{right}\). Curiously, these two approaches result in slightly different values for \(\bar{c}_0/c_\mathrm{source}\). I don’t understand the cause for this difference, but since \(N_\mathrm{right}\) is what has actually been measured, we use these values to estimate \(\bar{c}_0.\) The resulting equilibrium concentrations are
Test
\(N_\mathrm{right}\) (10-10 mol)
\(\bar{c}_0/c_\mathrm{source}\) (-)
1.3/0.01
4.10
0.038
1.3/0.05
10.2
0.097
1.3/0.1
17.8
0.168
1.3/0.4
41.4
0.395
1.3/1.0
52.4
0.445
1.6/0.01
1.21
0.014
1.6/0.05
3.64
0.043
1.6/0.1
6.15
0.072
1.6/0.4
13.0
0.154
1.6/1.0
21.6
0.225
1.9/0.01
0.41
0.006
1.9/0.05
1.14
0.018
1.9/0.1
1.64
0.025
1.9/0.4
3.19
0.051
1.9/1.0
8.19
0.113
We have now investigated two independent estimations of the chloride equilibrium concentrations: from mass balance of chloride tracers in the out-diffusion stage, and from measured stable chloride content. Here are plots comparing these two estimations
The similarity is quite extraordinary! With the exception of two
samples (1.3/0.4 and 1.9/0.1), the equilibrium chloride concentrations
evaluated in these two very different ways are essentially the
same. This result strongly confirms that the evaluations are adequate.
Steady-state fluxes
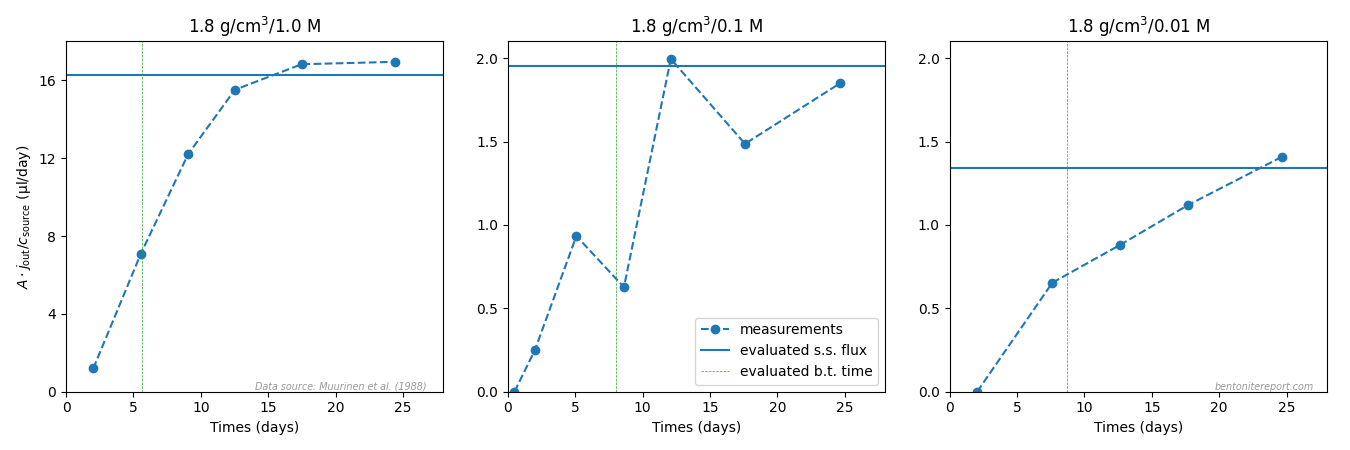
Vl07 present the flux evolution in the through-diffusion stage only for a single test (1.6/1.0), and it looks like this (left diagram)
The outflux reaches a relatively stable value after about 7 days,
after which it is meticulously monitored for a quite long time period.
The stable flux is not completely constant, but decreases slightly
during the course of the test. We anyway refer to this part as the
steady-state phase, and to the preceding part as the transient phase.
One reason that the steady-state is not completely stable is, reasonably, that the source reservoir concentration slowly decreases during the course of the test. The estimated drop from this effect, however, is only about one percent,8 while the recorded drop is substantially larger, about 7%. Vl07 do not comment on this perhaps unexpectedly large drop, but it may be caused e.g. by the ongoing conversion of the bentonite to a purer sodium state (see above).
Most of the analysis in Vl07 is based on anyway assigning a single
value to the steady-state flux. Judging from the above plot, Vl07 seem
to adopt the average value during the steady-state phase, and it is
clear that the assigned value is well constrained by the measurements
(the drop is a second order effect). The steady-state flux can
therefore be said to be directly measured in the through-diffusion
stage, rather than being obtained from fitting a certain model to
data.
Vl07 only implicitly consider the steady-state flux, in terms of a fitted “effective diffusivity” parameter, \(D_e\) (more on this in the next section). We can, however, “de-derive” the corresponding steady-state fluxes using \(j_\mathrm{ss} = D_e\cdot c_\mathrm{source}/L\), where \(L\) (= 0.01 m) is sample length. When comparing different tests it is convenient to use the normalized steady state flux \(\widetilde{j}_\mathrm{ss} = j_\mathrm{ss}/c_\mathrm{source}\), which then relates to \(D_e\) as \(\widetilde{j}_\mathrm{ss} = D_e/L\). Indeed, “effective diffusivity” is just a scaled version of the normalized steady-state flux, and it makes more sense to interpret it as such (\(D_e\) is not a diffusion coefficient). From the reported values of \(D_e\) we obtain the following normalized steady-state fluxes (my apologies for a really dull table)
Test
\(D_e\) (10-12 m2/s)
\(\widetilde{j}_\mathrm{ss}\) (10-10 m/s)
1.3/0.01
2.6
2.6
1.3/0.05
7.5
7.5
1.3/0.1
16
16
1.3/0.4
25
25
1.3/1.0
49
49
1.6/0.01
0.39
0.39
1.6/0.05
1.1
1.1
1.6/0.1
2.3
2.3
1.6/0.4
4.6
4.6
1.6/1.0
10
10
1.9/0.01
0.033
0.033
1.9/0.05
0.12
0.12
1.9/0.1
0.24
0.24
1.9/0.4
0.5
0.5
1.9/1.0
1.2
1.2
Plotting \(\widetilde{j}_\mathrm{ss}\) as a function of background concentration gives the following picture
The steady-state flux show a very consistent behavior: for all three
densities, \(\widetilde{j}_\mathrm{ss}\) increases with background
concentration, with a higher slope for the three lowest background
concentrations, and a smaller slope for the two highest background
concentrations. Although we have only been able to investigate the
1.6/1.0 test in detail, this consistency confirms that the
steady-state flux has been reliably determined in all tests.
Transient phase evaluations
So far, we have considered estimations based on more or less direct
measurements: stable chloride concentration profiles, tracer mass
balance in the out-diffusion stage, and steady-state fluxes. A major
part of the analysis in Vl07, however, is based on fitting solutions
of the diffusion equation to the recorded flux.
Vl07 state somewhat different descriptions for the through- and
out-diffusion stages. For out-diffusion they use an expression for the
flux into the right side reservoir (the sample is assumed located
between \(x=0\) and \(x=L\))
where \(j_\mathrm{ss}\) is the steady-state flux,9 \(D_e\) is “effective diffusivity”, and \(\epsilon_\mathrm{eff}\) is the effective porosity parameter (Vl07 also state a similar expression for the diffusion into the left side reservoir, but these results are discarded, as discussed earlier). For through-diffusion, Vl07 instead utilize the expression for the amount tracer accumulated in the right side reservoir
were \(S\) denotes the cross section area of the sample.
It is clear that Vl07 use \(D_e\) and \(\epsilon_\mathrm{eff}\) as fitting parameters, but not exactly how the fitting was conducted. \(D_e\) seems to have been determined solely from the the through-diffusion data, while separate values are evaluated for \(\epsilon_\mathrm{eff}\) from the through- and out-diffusion stages. As already discussed, Vl07 also provide a third estimation of \(\epsilon_\mathrm{eff}\), based on mass-balance in the out-diffusion stage. To me, the study thereby gives the incorrect impression of providing a whole set of independent estimations of \(\epsilon_\mathrm{eff}\). Although eqs. 5 and 6 are fitted to different data, they describe diffusion in one and the same sample, and an adequate fitting procedure should provide a consistent, single set of fitted parameters \((D_e, \epsilon_\mathrm{eff})\). Even more obvious is that the estimation of \(\epsilon_\mathrm{eff}\) from fitting eq. 5 should agree with the estimation from the mass-balance in the out diffusion stage — the accumulated amount in the right side reservoir is, after all, given by the integral of eq. 5. A significant variation of the reported fitting parameters for the same sample would thus signify internal inconsistency (experimental- or modelwise).
In the following reevaluation we streamline the description by solely using fluxes as model expressions,4 and by emphasizing steady-state flux as a parameter, which I think gives particularly neat expressions,10 (“TD” and “OD” denote through- and out-diffusion, respectively)
Here we use the pore diffusivity, \(D_p\), instead of the combination \(D_e/\epsilon_\mathrm{eff}\) in the exponential factors, and \(\widetilde{j} = j/c_\mathrm{source}\) denotes normalized flux. This formulation clearly shows that the time evolution is governed solely by \(D_p\), and that \(\widetilde{j}_\mathrm{ss}\) simply acts as a scaling factor.
In my opinion, using \(\widetilde{j}_\mathrm{ss}\) and \(D_p\) gives a formulation more directly related to measurable quantities; the steady-state flux is directly accessible experimentally, as we just examined, and \(D_p\) is an actual diffusion coefficient (in contrast to \(D_e\)) that can be directly evaluated from clay concentration profiles. Of course, eqs. 7 and 8 provide the same basic description as eqs. 5 and 6, and \(\widetilde{j}_\mathrm{ss}\) and \(D_p\) are related to the parameters reported in Vl07 as
When reevaluating the reported data we focus on the above discussed consistency aspect, i.e. whether or not a single model (a single pair of parameters) can be satisfactory fitted to all available data for the same sample. In this regard, we begin by noting that the fitting parameters are already constrained by the direct estimations. We have already concluded that the recorded steady-state flux basically determines \(\widetilde{j}_\mathrm{ss}\), and if we combine this with the estimated chloride clay concentration, \(D_p\) is determined from \(j_\mathrm{ss} = \phi\cdot D_p\cdot \bar{c}_0/L\), i.e.
Here are plotted values of \(D_p\) evaluated in this manner
Note that these values basically remain constant for samples of similar density (within a factor of 2) as the background concentration is varied by two orders of magnitude. This is the expected behavior of an actual diffusion coefficient,11 and confirms the adequacy of the evaluation; the numerical values also compares rather well with corresponding values for “MX-80” bentonite, measured in closed-cell tests (indicated by dashed lines in the figure).
Using eq. 10, we can also evaluate values of \(D_p\) corresponding to
the various reported fitted parameters \(\epsilon_\mathrm{eff}\). The
result looks like this (compared with the above evaluations from
direct estimations)
As pointed out above, a consistent evaluation requires that the
parameters fitted to the out-diffusion flux (red) are very similar
to those evaluated from considering the mass balance in the same process
(blue). We note that the resemblance is quite reasonable, although
some values — e.g. tests 1.3/1.0 and 1.6/1.0 — deviate in a perhaps
unacceptable way.
\(D_p\) evaluated from reported through-diffusion parameters, on the other hand, shows significant scattering (green). As the rest of the values are considerably more collected, and as the steady-state fluxes show no sign whatsoever that the diffusion coefficient varies in such erratic manner, it is quite clear that this scattering indicates problems with the fitting procedure for the through-diffusion data.
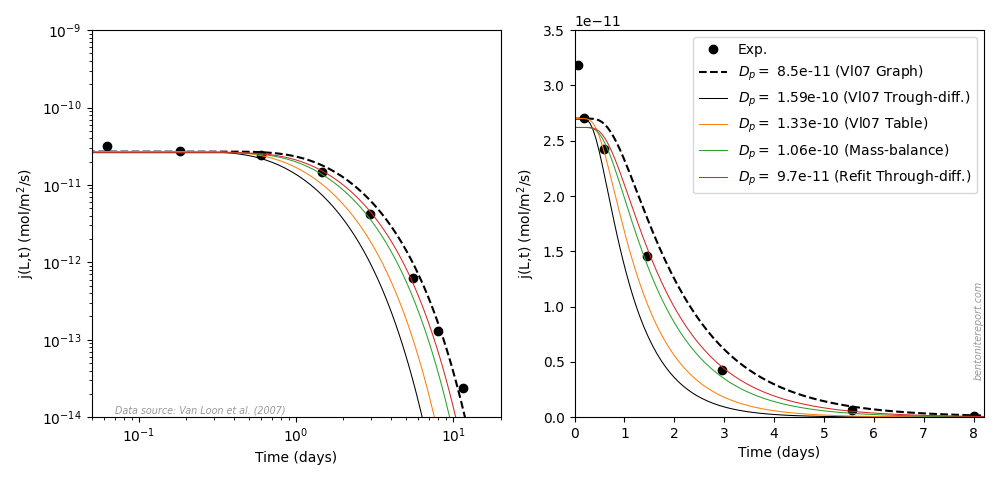
The 1.6/1.0 test
To further investigate the fitting procedures, we take a detailed look at the 1.6/1.0 test, for which flux data is provided. Vl07 report fitted parameters \(D_e = 1.0\cdot 10^{-11}\) m2/s and \(\epsilon_\mathrm{eff} = 0.063\) to the through-diffusion data, corresponding to \(\widetilde{j}_\mathrm{ss} = 1.0\cdot 10^{-9}\) m/s and \(D_p = 1.6\cdot 10^{-10}\) m2/s. We have already concluded that the steady-state flux is well captured by this data, but to see how well fitted \(\epsilon_\mathrm{eff}\) (or \(D_p\)) is, lets zoom in on the transient phase
This diagram also contains models (eq. 7) with different values of \(D_p\), and with a slightly different value of \(j_\mathrm{ss}\).12 It is clear that the model presented in the paper (black) completely misses the transient phase, and that a much better fit is achieved with \(D_p = 9.7\cdot10^{-11}\) m2/s (and \(\widetilde{j}_\mathrm{ss} = 1.06\cdot 10^{-9}\) m/s) (red). This difference cannot be attributed to uncertainty in the parameter \(D_p\) — the reported fit is simply of inferior quality. With that said, we note that all information on the transient phase is contained within the first three or four flux points; the reliability could probably have been improved by measuring more frequently in the initial stage.13
A reason for the inferior fit may be that Vl07 have focused only on the linear part of eq. 6; the paper spends half a paragraph discussing how the approximation of this expression for large \(t\) can be used to extract the fitting parameters using linear regression. Does this mean that only experimental data for large times where used to evaluate \(D_e\) and \(\epsilon_\mathrm{eff}\)? Since we are not told how fitting was performed, we cannot answer this question. Under any circumstance, the evidently low quality of the fit puts in question all the reported \(\epsilon_\mathrm{eff}\) values fitted to through-diffusion data. This is actually good news, as several of the corresponding \(D_p\) values were seen to be incompatible with constraints from direct estimations. We can thus conclude with some confidence that the inconsistency conveyed by the differently evaluated fitting parameters does not indicate experimental shortcomings, but stems from bad fitting of the through-diffusion model. Therefore, we simply dismiss the reported \(\epsilon_\mathrm{eff}\) values evaluated in this way. Note that the re-fitted value for \(D_p\) \((9.7\cdot10^{-11}\) m2/s) is consistent with those evaluated from direct estimations.
We note that when fitting the transient phase, it is appropriate to
use a value of \(\widetilde{j}_\mathrm{ss}\) slightly larger than the
average value adopted by Vl07 (as the model does not account for the
observed slight drop of the steady-state flux). This is only a minor
variation in the \(\widetilde{j}_\mathrm{ss}\) parameter itself (from
\(1.02\cdot10^{-9}\) to \(1.06\cdot10^{-9}\) m/s), but, since this value
sets the overall scale, it indirectly influences the fitted value of
\(D_p\) (model fitting is subtle!).
More questions arise regarding the fitting procedures when also examining the presented out-diffusion stage for the 1.6/1.0 sample. The tabulated fitted value for this stage is \(\epsilon_\mathrm{eff}\) = 0.075, while it is implied that the same value has been used for \(D_e\) as evaluated from the the through-diffusion stage (\(1.0\cdot 10^{-11}\) m2/s). The corresponding pore diffusivity is \(D_p = 1.33\cdot 10^{-10}\) m2/s. The provided plot, however, contains a different model than tabulated, and looks similar to this one (left diagram)
Here the presented model (black dashed line) instead corresponds to
\(D_p = 8.5\cdot 10^{-11}\) m2/s (or \(\epsilon_\mathrm{eff}\) =
0.118). The model corresponding to the tabulated value (orange) do not
fit the data! I guess this error may just be due to a typo in the
table, but it nevertheless gives more reasons to
not trust the reported \(\epsilon_\mathrm{eff}\) values fitted to
diffusion data.
The above diagram also shows the model corresponding to the reported parameters from the through-diffusion stage (black solid line). Not surprisingly, this model does not fit the out-diffusion data, confirming that it does not appropriately describe the current sample. The model we re-fitted in the through-diffusion stage (red), on the other hand, captures the outflux data quite well. By also slightly adjusting \(\widetilde{j}_{ss}\), from from \(1.06\cdot10^{-9}\) to \(0.99\cdot10^{-9}\) m/s, to account for the drop in steady-state flux during the course of the through-diffusion test, and by plotting in a lin-lin rather than a log-log diagram, the picture looks even better! In a lin-lin plot (right diagram), it is easier to note that the model presented in the graph of Vl07 actually misses several of the data points. Could it be that Vl07 used visual inspection of the model in a log-log diagram to assess fitting quality? If so, data points corresponding to very low fluxes are given unreasonably high weight.14 This could be (another) reason for the noted difference between \(D_p\) evaluated from fitted parameters to the out-diffusion flux, and from the total accumulated amount of tracer (which should be equal).
From examining the reported results of sample 1.6/1.0 we have seen that the fitting procedures adopted in Vl07 appear inappropriate, but also that a consistent model can be successfully fitted to all available data (using a single \(D_p\)). Vl07 don’t provide flux data for any other sample, but we must conclude that the reported fitted \(\epsilon_\mathrm{eff}\) parameters cannot be trusted. Luckily, the preformed refitting exercise confirms the results obtained from analysis of stable chloride profiles and accumulated amount of tracers in out-diffusion, and we conclude that these results most probably are reliable. The corresponding value of \(\bar{c}_0/c_\mathrm{source}\) (using eq. 11) for the refitted model is here compared with the estimations from direct measurements
Summary and verdict
Chloride equilibrium concentrations evaluated from mass balance of the tracer in the out-diffusion stage and from stable chloride content show remarkable agreement. On the other hand, the scattering of estimated concentrations increases substantially if they are also evaluated from the reported fitted diffusion parameters. This could indicate underlying experimental problems, as a consistent evaluation should result in a single value for the equilibrium concentration; the various evaluations — stable chloride, out-diffusion mass balance, through-diffusion fitting and out-diffusion fitting — relate, after all, to a single sample.
By reexamining the evaluations we have found, however, that the problem is associated with how the fitting to diffusion data has been conducted (and presented), rather than indicating fundamental experimental issues. In the test that we have been able to examine in detail (1.6/1.0), we found that the reported models do not fit data, but also that it is possible to satisfactorily refit a single model that is also compatible with the direct methods for evaluating the equilibrium concentration. For the rest of the samples, we have also been able to discard the fitted diffusion parameters, as they are not compatible e.g. with how the steady-state flux (very consistently) vary with density and background concentration.
For these reasons, we discard the reported “effective porosity”
parameters evaluated from fitting solutions of the diffusion equation
to flux data, and keep the results from direct measurements of
chloride equilibrium concentrations (from stable chloride profile
analysis and mass-balance in the out-diffusion stage). I judge the
resulting chloride equilibrium concentrations as reliable and that
they can be used for increased qualitative process understanding. I
furthermore judge the directly measured steady-state fluxes as
reliable. This study thus provide adequate values for both chloride
equilibrium concentrations and diffusion coefficients.
However, a frustrating problem is that, although the equilibrium concentrations are well determined, we have little information on the exact state of the samples in which they have been measured. We basically have to rely on that the “KWK” material is “similar” to “MX-80”, keeping in mind that “MX-80” is not really a uniform material (from a scientific point of view). Also, the exchangeable mono/divalent cation ratio is most probably quite different in samples contacted with different background concentrations.
Yet, I judge the present study to provide the best information
available on chloride equilibrium in compacted bentonite, and will use
it e.g. for investigating the salt exclusion mechanism in these
systems (Ialreadyhave). That this information is the best available is, however, also
a strong argument for that more and better constrained data is
urgently needed.
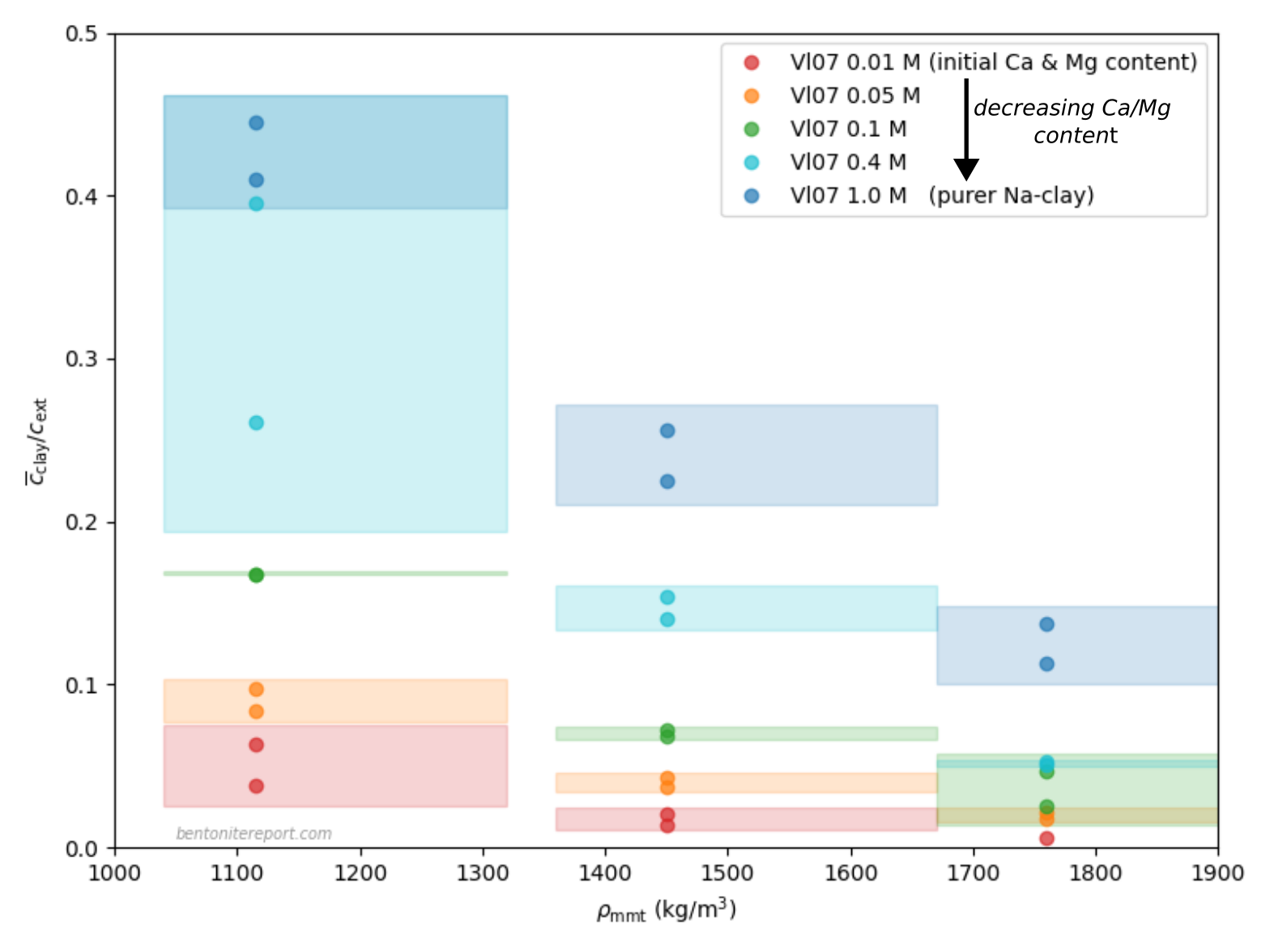
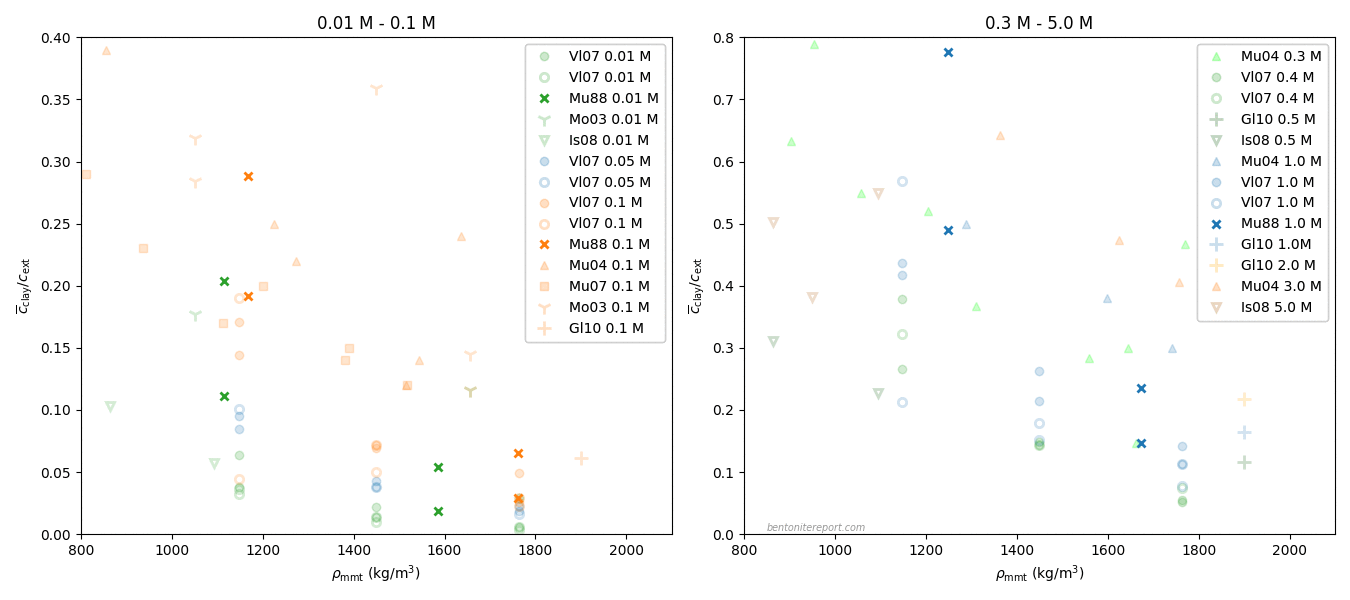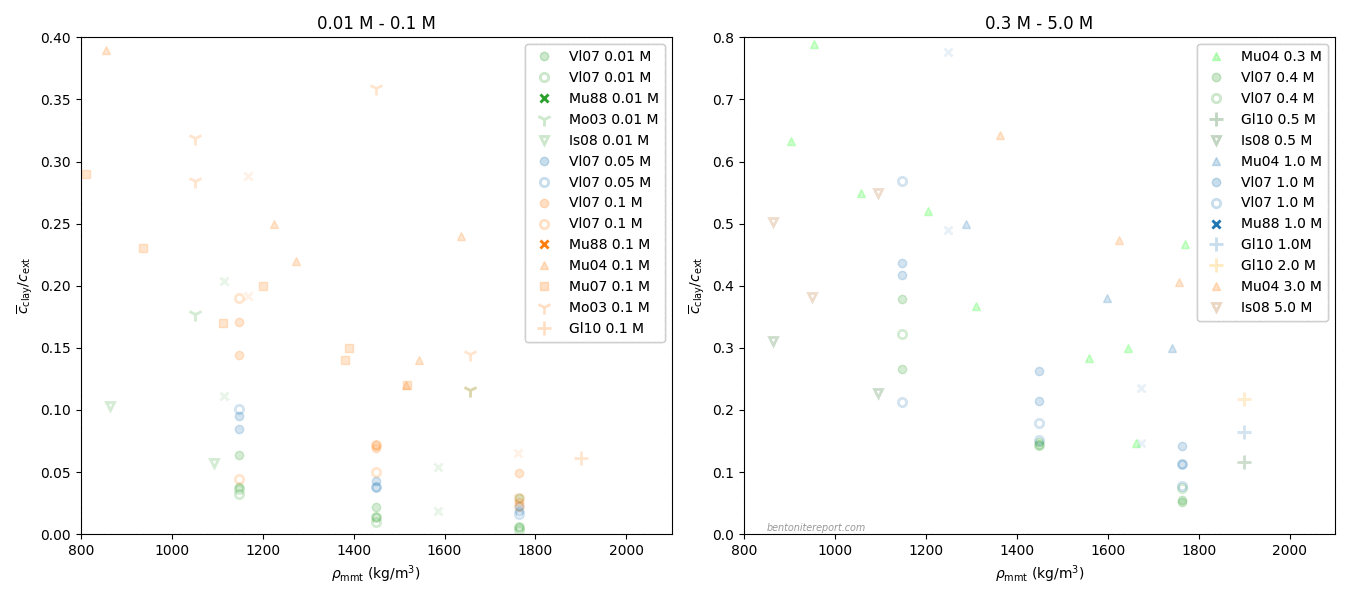
The (reliable) results are presented in the diagram below, which includes “confidence areas”, that takes into account the spread in equilibrium concentrations, in samples where more than a single evaluation were performed, and the estimated uncertainty in effective montmorillonite dry density (the actual points are plotted at nominal density, assuming 80% montmorillonite content)
[1] Vejsada et al. (2006) call their material “KWK 20-80”. In other
contexts, I have also found the versions
“KWK
food grade” and
“KWK krystal klear”. I have given up my attempts at trying to
understand the difference between these “KWK” variants.
[3] This should be relatively straightforward, but I get at bit nervous e.g. about the presence of a rather arbitrary factor 0.85 in the presented formula (eq. 19 in Van Loon et al. (2007)).
[4] As always for these types of diffusion tests, the raw data consists of simultaneously measured values of time (\(\{t_i\}\)) and reservoir concentrations (\(\{c_i\}\)). From these, flux can be evaluated as (\(A\) is sample cross sectional area, and \(V_\mathrm{res}\) is reservoir volume)
\(\bar{j}_i\) is the mean flux in the time interval between \(t_{i-1}\)
and \(t_i\), and should be associated with the average time of the
same interval: \(\bar{t}_i = (t_i + t_{i-1})/2\). The above formula
assumes no solution replacement after the \((i-1)\):th measurement (if
the solution is replaced, \(\left (c_i – c_{i-1} \right )\) should be
replaced with \(c_i\)).
Alternatively one can work with the accumulated amount of substance, which e.g. is \(N(t_i) = \sum_{j=1}^i c_j\cdot V_\mathrm{res}\), in case the solution is replaced after each measurement. I prefer using the flux because eq. * only depends on two consecutive measurements, while \(N(t_i)\) in principle depends on all measurements up to time \(t_i\). Also, I think it is easier to judge how well e.g. a certain model fits or is constrained by data when using fluxes; the steady-state, for example, then corresponds to a constant value.
Van Loon et
al. (2007) seem to have utilized both fluxes and accumulated
amount of substance in their evaluations, as discussed in later
sections.
[8] From total test time, recorded flux, and sample cross sectional area, we estimate that about \(5.8\cdot 10^{-8}\) mol of tracer is transferred from the source reservoir during the course of the test (\(50\) days\(\cdot 2.7\cdot 10^{-11}\) mol/m2/s\(\cdot 0.0005\) m2). This is about 1% of the total amount tracer, \(c_\mathrm{source} \cdot V_\mathrm{source} = 2.65 \cdot 10^{-5}\) M \(\cdot 0.2\) L = \(5.3\cdot 10^{-6}\) mol.
[9] Van Loon et al. (2007) label this parameter \(J_L\), and don’t relate it explicitly to the steady-state flux. From the experimental set-up it is clear, however, that the initial value of the out-diffusion flux (into the right side reservoir) is the same as the previously maintained steady-state flux. Note that the expressions for the fluxes in the out-diffusion stage in Van Loon et al. (2007) has the wrong sign.
[10] The description provided by eqs. 5 and 6 not only mixes expressions for flux and accumulated amount tracer, but also contains three dependent parameters \(D_e\), \(\epsilon_\mathrm{eff}\), and \(j_\mathrm{ss}\) (e.g. \(j_\mathrm{ss} = D_e/(c_\mathrm{source}\cdot L)\)). In this reformulation, the model parameters are strictly only \(\widetilde{j}_\mathrm{ss}\) and \(D_p\). We have also divided out \(c_\mathrm{source}\) to obtain equations for normalized fluxes. Note that the expression for \(\widetilde{j}_{TD}(L,t)\) is essentially the same that we have used in previousassessments of through-diffusion tests. Note also that eqs. 7 and 8 imply the relation \(\widetilde{j}_{OD}(L,t) = \widetilde{j}_{ss} – \widetilde{j}_{TD}(L,t)\), reflecting that the out-diffusion process is essentially the through-diffusion process in reverse.
[11] Note the similarity with that diffusivity also is basically independent of background concentration for simple cations. Note also that there is no reason to expect completely constant \(D_p\) for a given density, because the samples are not identically prepared (being saturated with saline solutions of different concentration).
[12] As we here consider a single sample, we alternate a bit sloppily between steady-state flux (\(j_\mathrm{ss} \)) and normalized steady-state flux (\(\widetilde{j}_\mathrm{ss}\)), but these are simply related by a constant: \(\widetilde{j}_\mathrm{ss} = j_\mathrm{ss} / c_\mathrm{source}\). For the 1.6/1.0 test this constant is (as tabulated) \(c_\mathrm{source} = 2.65\cdot 10^{-2}\) mol/m3.
[13] I think it is a bit amusing that the pattern of data points suggests measurements being performed on Mondays, Wednesdays, and Fridays (with the test started on a Wednesday).
[14] I have warned about the dangers of log-log plots earlier.
In contrast to the
earlierassessed
studies, Mu04 is not a diffusion study, but considers directly the
clay concentration in samples equilibrated with an external
solution. Moreover, Mu04 uses purified “MX-80” bentonite, ion
exchanged to a more pure sodium form.
Mu04 contains data from two quite different types of samples. 15
samples originate from a study on pressure response in montmorillonite
contacted with external NaCl solutions of varying concentration
(Karnland et al., 2005; in the following referred to as Ka05). The
remaining 10 samples were prepared for determining basal distance
using small-angle X-ray scattering (SAXS). We refer to these two sets
of samples as the swelling pressure samples and the SAXS
samples, respectively. A more detailed description of the sample
analysis is given in
Muurinen (2006), in the following referred to as Mu06.
Material
The used material is referred to as “purified MX-80”. Mu06 states that this material was produced by mixing “MX-80” powder and NaCl solutions in bottles, where the solutions were repeatedly replaced. Mu06 also states that “During this process, part of the dissolving accessory minerals was removed as well.” Ka05 more explicitly say that the raw material was “converted into a homo-ionic Na+ state and coarser grains were removed (Muurinen et al., 2002). The montmorillonite content was thereby increased to above 90% of the total material.”1 With no better estimate of the montmorillonite content, we therefore associate the stated densities with effective montmorillonite dry density, i.e. we assume a montmorillonite content of 100%. We should keep in mind the uncertainty of this parameter, and that, reasonably, this choice somewhat overestimates the effective montmorillonite dry density.
The purified material was found to leach sulfate and carbonate,
indicating that it still contains some amount of soluble accessory
minerals. It follows that the montmorillonite is not completely of
pure sodium form, as confirmed by the reported exchangeable ion
population: 0.74 eq/kg sodium, 0.06 eq/kg calcium, and 0.03 eq/kg
magnesium (i.e. a di/mono-valent ratio of about 10/90). It is
interesting that the material still contains a non-negligible amount
of divalent ions, given that quite a lot of effort was put into
producing it. Nevertheless, we can assume that this material contains
considerably more sodium as compared with the “raw” “MX-80”
encountered in the
previouslyassessed
studies.
Samples overview
The swelling pressure samples were originally cylindrical, with diameter 5 cm and length 2 cm, giving a volume of approximately 39 cm3. After termination of the swelling pressure tests, these samples were cut into pieces, to be used for different types of analyses. The samples cover large ranges of density and external NaCl concentration, as listed here
Neither Mu04 nor Mu06 provide much information about preparation and handling of the SAXS samples. It is stated that these are cylindrical with diameter 2.5 cm and length 0.5 cm, giving a total volume of 2.45 cm3. Although not stated, also these sample have reasonably been sub-divided, as e.g. density was determined (and some parts were obviously used for SAXS).
The SAXS samples varies substantially in density, but were only contacted with external NaCl solutions of concentration 0.1 or 0.3 M. The table below identifies each sample by external solution concentration and, presumably, measured density (how density was determined is not reported)
NaCl conc. (M)
\(\rho_d\) (measured?) (g/cm3)
0.1
0.750
0.1
0.875
0.1
1.225
0.1
1.516
0.1
1.543
0.3
0.954
0.3
1.058
0.3
1.206
0.3
1.559
0.3
1.662
In the following, we separately discuss the chloride concentration
evaluations of the swelling pressure samples and the SAXS samples.
Swelling pressure samples
Chloride concentration was
evaluated3 in three separate pieces of each original sample, as
indicated in this figure:4
Chloride content was determined by dispersing each piece, containing about 1 g of clay, in de-ionized water, centrifuging, and analyzing the supernatant. The pieces were located at different heights of the original cylinder (see figure), giving some spatial resolution of the chloride distribution, reported in Mu06. Mu04, however, only report the average value for each sample. For some samples, the value reported in Mu04 does not perfectly match the average calculated from the values listed in Mu06 (cf. the plots below).
Let’s anticipate the “verdict” for these samples: the evaluated clay
concentrations are not useful for quantitative understanding of
ion equilibrium, and I will not use them e.g. for validating anion
exclusion models.
That these samples are not adequately equilibrated is best seen from
looking at the evaluated concentrations in the samples contacted with
0.1 and 0.3 M NaCl, here plotted with spatial resolution
The indicated densities (\(\rho_d\)) are the average of the spatially resolved values reported in Mu06 (these differ a bit from what is reported in Mu04). The profiles show several peculiarities:
The densest samples in both test sets (S2-18 and S2-16, respectively) contain the second highest amount of chloride.
Samples S2-21 and S2-02 have a huge difference in chloride concentration, even though they have quite similar density.
In both test sets, the chloride concentration is very similar in the samples with densities \(\sim\) 1.3 g/cm3 and \(\sim\) 1.6 g/cm3 (S2-04 vs. S2-17, and S2-14 vs. S2-15).
These observations strongly indicate that the samples either have not
been adequately equilibrated, or that they have not been adequately
handled after test termination (or both). Consequently, the results
are of little help for adequate quantitative process
understanding.5 Mu04 acknowledge this shortcoming,
but takes different action
At high dry densities (>1630 kg/m3 ) and low NaCl concentrations, the concentrations in the porewater tend to increase with increasing density. The phenomenon is not seen with the thinner SAXS samples, however. One possible explanation is that during saturation too much chloride is transported into the sample and the equilibration time has been too short to reach the equilibrium. Three such samples marked with (*) in Tables 1 and 3 have been omitted from the treatment of the results.
But one cannot simply omit only the samples that deviate from the
expected qualitative behavior while assuming that the rest of the
results are adequate! This is especially true when the source for the
shortcoming has not been clarified. In fact, we just identified
additional peculiarities in the data. Consequently, not only should
the rest of the samples equilibrated with 0.1 M and 0.3 M NaCl be
omitted, but also those equilibrated with 1.0 M and 3.0 M.
For completeness, here are the chloride concentration profiles for
the tests with high background concentration
Although we discard them, it may be interesting to identify possible
reasons for these flawed results. Previously, we discussed why
equilibrium salt concentrations may be overestimated. Both factors
identified there may apply here: failing to handle possible interface
excess, and issues related to directly saturating samples with a
saline solution.
Saturating with saline solutions
From the different reports it is clear that the samples were saturated directly with the saline solution. It is, however, not fully clear if the saturation was performed from only one end of the sample, or from both. In Mu04 and Mu06, the assumption seems to be that the samples were saturated from one side only, although this is not described in any detail. Mu04 write
The compacted samples were closed in metal tubes and saturated
through a sinter at one end.
In Ka05, however, the statement is
The water solutions were slowly circulated behind the bottom filters
to start with, in order not to trap the original air in the samples.
where the formulation “to start with” suggests that the solution was
eventually also contacted from the top.
The reason why this detail may be important is that with solution at
one end only the effective diffusion distance for salt is doubled as
compared with having solution at both ends. A doubling of diffusion
length, in turn, increases the characteristic diffusion time by a
factor of 4.
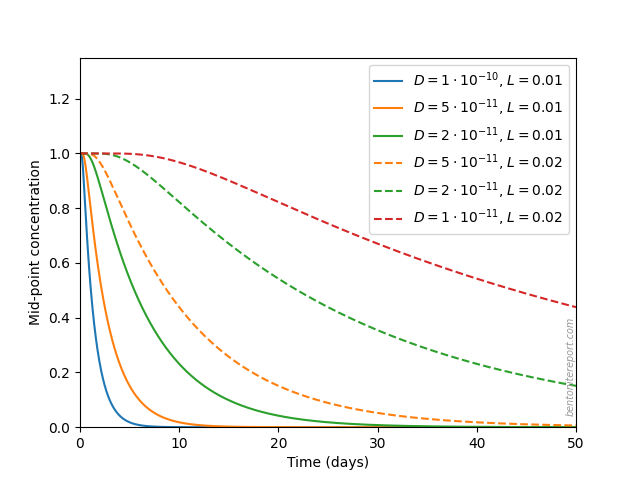
We estimate the time needed for excess salt to diffuse out by
considering a model with an initial unit concentration in the entire
domain (domain length \(L\)), and boundary condition of zero
concentration at the end points. The midpoint concentration in such a
model, for various values of \(L\) (0.01 — 0.02 m) and diffusion
coefficients (\(1\cdot 10^{-10}\) — \(1\cdot 10^{-11}\;\mathrm{m^2/s}\)),
evolve like this
We see that, depending on parameter values, the set-up may be such that a possible “overshoot” of salt have not had time to completely diffuse out of the sample during the course of the swelling pressure tests, which were conducted for about a month. In particular, if the effective diffusion length was 2 cm during the major part of the saturation process, it is very plausible that the equilibrium process was not completed for certain samples (this depends of course also on the detailed values of diffusion coefficient and equilibration time).
Supported by this simple analysis, we cannot rule out that the samples
initially took up more salt than dictated by the final state, and that
this salt may not have had time to fully diffuse out again.
Interface excess
Concerning interface excess (a potential problem regardless of whether or not the sample has reached full equilibrium before termination), no detailed information is given on the dismantling procedure. It seems relatively clear, though, that the outer parts of the original samples were not sectioned off. Ka05 write
After reaching pressure equilibrium and a minimum test time of 1
month, the test solutions were disconnected and the samples were
removed and split in order to make detailed analyses of the water
ratio, sample density, pore-water chemistry, water activity and
microstructure.
Mu06 writes (“Figure 4” is similar to the figure above)
The bentonite sample cylinders obtained from the swelling pressure
measurements were cut into smaller pieces according to Figure 4 in
order to provide samples for different analyses and
measurements. Half of the sample piece was used for the porewater
studies while the other half […] was left in Clay Technology AB
for their studies.
My interpretation is that the original sample was cut in half during the dismantling in the swelling pressure study, and that one half was sent off elsewhere for the analysis presented in Mu04 (the upper part of the disc indicated in the above figure). Thus, it seems plausible that the interface regions were not sectioned off during dismantling, and that the samples were stored/transported for an appreciable amount of time. Possible excess salt would consequently had time to even out in the sample before further analysis. This interpretation is in line with the evaluated rather flat chloride profiles: note the contrast between these and the quite pronouncednon-linear profiles observed at the interfaces in studies where samples are sectioned at test termination.
SAXS samples
Mu04 (and Mu06) provide almost no information about how the SAXS samples were handled. Reasonably, also these samples were split, with some part being used for the SAXS measurement and another for determining water content, but we have no information on this. In fact, not even the SAXS results are properly reported for these samples; only evaluated “interlamellar spaces” for the samples equilibrated at 0.3 M are discussed; neither Mu04 nor Mu06 report SAXS data for the 0.1 M samples.
The reports are also somewhat contradictory. In the caption to a table in Mu04 it is stated that the SAXS samples were first saturated with de-ionized water, and thereafter equilibrated with the salt solutions. Mu06, on the other hand, states
The samples were compacted into the cells and saturated through a
filter plate from one side with 0.1 or 0.3 M NaCl solutions for 12
days.
Should the last statement rather be that equilibration was
performed for 12 days, after saturation? Under any
circumstance, the lack of information on handling of the SAXS samples
is a major flaw and must be considered in the assessment.
If I should guess, I believe that possible interface excess on these
samples where not handled, i.e. I believe that the end parts
were not sectioned off when the samples were dismantled (also, with
only 5 mm thick samples, there is not much to section off…). Note
that the SAXS samples are thin (5 mm) and were equilibrated with
solutions of relatively low concentration (0.1 M and 0.3 M). Based on
the
analysis in the previous post, these samples are expected to be very
sensitive to an interface excess effect.
Here is plotted the reported chloride clay concentrations for the SAXS
samples, together with corresponding (average) values for the swelling
pressure samples at the same background concentration6
Note that, although the density dependence on the SAXS sample data appears more reasonable compared with the swelling pressure samples, the SAXS sample data seem to have a scatter of at least a factor of 2 (see e.g. the leftmost SAXS points for 0.3 M and the rightmost points for 0.1 M). Note also that in one sample (0.1 M, 750 kg/cm3), the evaluated clay concentration is larger than the background concentration!
Summary and verdict
I discard the chloride concentrations measured in the swelling pressure samples, based on the reported results: it is clear that the observed scatter and spurious dependencies demonstrate that the samples were not properly equilibrated, in order to use the results for quantitative process understanding. To accept e.g. a result that the equilibrium chloride concentration increases with density I require a considerably more rigorous study. Moreover, I mean that all results must be discarded, not only those that obviously deviate from the expected qualitative behavior.
I also discard the results of the SAXS samples. Although we don’t have
any clear indication that they were incorrectly prepared, I judge the
uncertainties and lack of information to be too large in order to rely
on the results. Almost no information is provided! Furthermore, the
reports do not give any hint that the issue of interface excess is
identified and handled — an effect we can expect to be substantial in
these samples.
I am saddened to have to discard these results, because, in my mind, adequate results from equilibration of homo-ionic samples would be very valuable for increased process understanding. I strongly believe that the bentonite research community should strive for conducting many more of these relatively simple tests on purified clays, rather than complicated through-diffusion tests. In properly conducted equilibrium tests, concentration data is accessed directly and there is no risk for the results to be obscured by issues related to ionic transport.
[3] Muurinen et al. (2004) also report chloride concentrations from
so-called squeezing tests. Squeezing tests are not adequate for
evaluating equilibrium clay concentrations, and I intend to write a
future blog post on the subject. Here we simply ignore the squeezing
results.
[4] The pieces labeled “B” were used
to determine density (water content).
[6] The plots also show the difference in average concentration and density for the swelling pressure samples as reported in Muurinen et al. (2004) and Muurinen (2006); these points should lie on top of each other.
When discussing semi-permeability, we noted that a bentonite sample that is saturated with a saline solution probably contains more salt in the initial stages of the process than what is dictated by the final state Donnan equilibrium. This salt must consequently diffuse out of the sample before equilibrium is reached.
The reason for such a possible “overshoot” of the clay concentration is that an infiltrating solution is not subject to a Donnan effect (between sample and external solution) when it fills out the air-filled voids of an unsaturated sample. Also, even if the region near the interface to the external solution becomes saturated — so that a Donnan effect is active — a sample may still take up more salt than prescribed by the final state, due to hyperfiltration: with a net inflow of water and an active Donnan effect, salt will accumulate at the inlet interface (unless the interface is flushed). This increased concentration, in turn, alters the Donnan equilibrium at the interface, with the effect that more salt diffuses into the clay.
These effects are relevant for our ongoing assessment of studies of chloride equilibrium concentrations. If bentonite samples are saturated with saline solutions, without taking precautions against these effects, evaluated equilibrium concentrations may be overestimated. Note that, even if saturating a sample may be relatively fast, it may take a long time for salt to reach full equilibrium, depending on details of the experimental set-up. In particular, if the set-up is such that the external solution does not flow past the inlet, equilibration may take a very long time, being limited by diffusion in filters and tubing.
Interface excess salt
Another way for evaluated salt concentrations to overestimate the true equilibrium value — which is independent of whether or not the sample has been saturated with a saline solution — is due to excess salt at the sample interfaces.
Suppose that you determine the equilibrium salt concentration in a bentonite sample in the following way. First you prepare the sample in a test cell and contact it with an external salt solution via filters. When the system (bentonite + solution) has reached equilibrium (taking all the precautions against overestimation discussed above), the concentration profile may be conceptualized like this
The aim is to determine \(\bar{c}_\mathrm{clay}\), the
clay concentration of the species of interest
(e.g. chloride), and to relate it to the corresponding concentration in the
external solution (\(c_ \mathrm{ext}\)).
After ensuring the value of \(c_\mathrm{ext}\) (e.g. by sampling or controlling the external solution), you unload the test cell and isolate the bentonite sample. In doing so, we must keep in mind that the sample will begin to swell as soon as the force on it is released, if only water is available. In the present example it is difficult not to imagine that some water is available, e.g. in the filters.1
It is thus plausible that the actual concentration profile look
something like this directly after the sample has been isolated
We will refer to the elevated concentration at the interfaces as the
interface excess. The exact shape of the resulting
concentration profile depends reasonably on the detailed procedure for
isolating the sample.2 If the ion content of the sample is measured
as a whole, and/or if the sample is stored for an appreciable amount
of time before further analysis (so that the profile evens out due to
diffusion), it is clear that the evaluated ion content will be larger
than the actual clay concentration.
To quantify how much the clay concentration may be overestimated due
to the interface excess, we introduce an effective penetration
depth, \(\delta\)
\(\delta\) corresponds to a depth of the external concentration that
gives the same interface excess as the actual distribution. Using this
parameter, it is easy to see that the clay concentration evaluated as
the average over the entire sample is
This expression is quite interesting. We see that the relative
overestimation, reasonably, depends linearly on \(\delta\) and on the
inverse of sample length. But the expression also contains the ratio
\(r \equiv c_\mathrm{ext}/\bar{c}_\mathrm{clay}\), indicating that the effect may
be more severe for systems where the clay concentration is small in
comparison to the external concentration (high density, low
\(c_\mathrm{ext}\)).
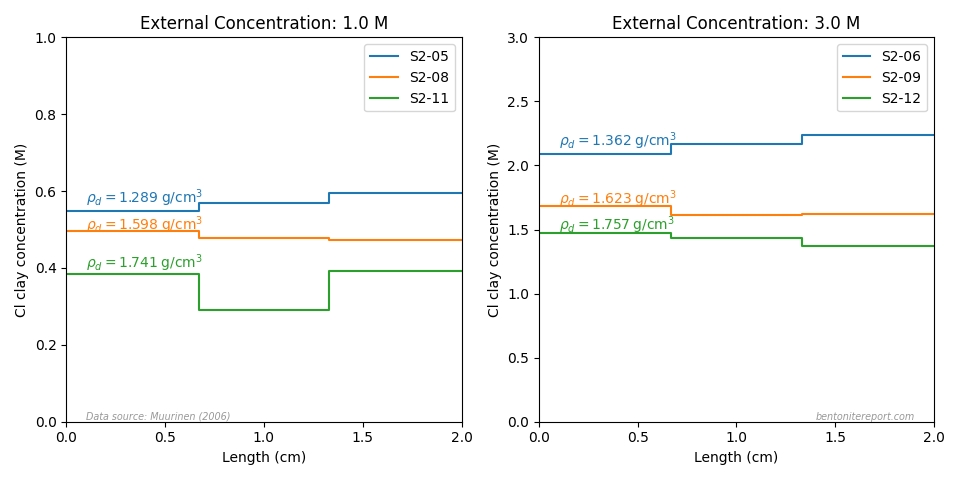
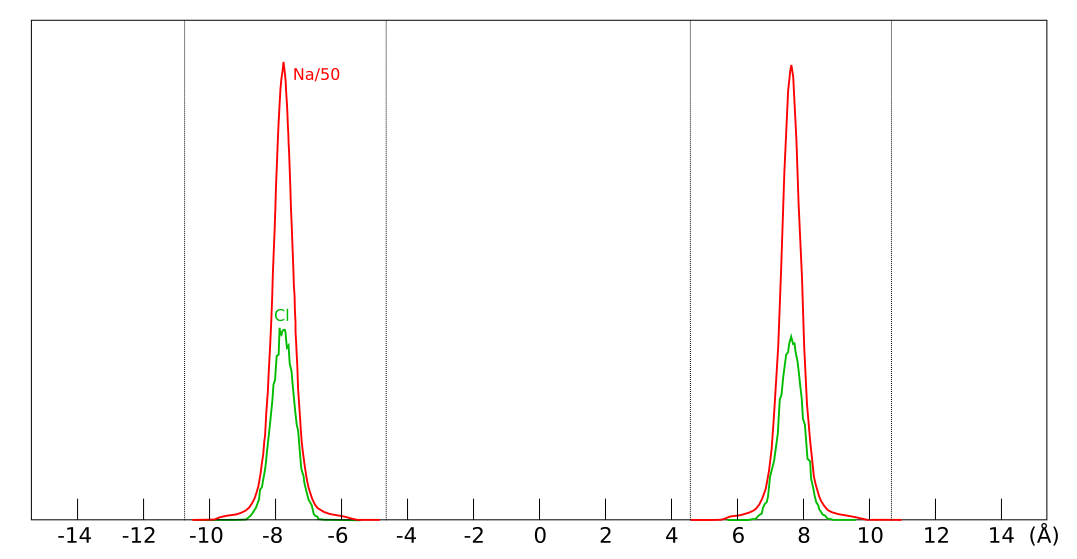
An interface excess is more than a theoretical concept, and is frequently observed e.g. in anion through-diffusion studies. We have previously encountered them when assessing the diffusion studies of Muurinen et al. (1988) and Molera et al. (2003).3Van Loon et al. (2007) clearly demonstrate the phenomenon, as they evaluate the distribution of stable chloride (the background electrolyte) in the samples after performing the diffusion tests.4 Here is an example of the chloride distribution in a sample of density 1.6 g/cm3 and background concentration of 0.1 M5
The line labeled \(\bar{c}_\mathrm{clay}\) is evaluated from the average of only the interior sections (0.0066 M), while the line labeled \(\bar{c}_\mathrm{eval}\) is the average of all sections (0.0104 M). Using the full sample to evaluate the chloride clay concentration thus overestimates the value by a factor 1.6. From eq. 1, we see that this corresponds to \(\delta = 0.2\) mm. For a sample of length 5 mm with the same penetration depth, the corresponding overestimation is a factor of 2.1.
Here is plotted the relative overestimation (eq. 1) as a function of \(\delta\) for several systems of varying length and \(r\) (\(= c^\mathrm{ext}/\bar{c}_\mathrm{clay}\))
We see that systems with large \(r\) and/or small \(L\) become
hypersensitive to this effect. Thus, even if it may be expected that
\(\delta\) decreases with increasing \(r\)6, we may still expect an
increased overestimation for such systems.
To avoid this potential overestimation of the clay concentration, I
guess the best practice is to quickly remove the first couple of
millimeters on both sides of a sample after it has been unloaded. In
many through-diffusion tests, this is done as part of the study, as
the concentration profile across the sample often is measured. In
studies where samples are merely equilibrated with an external
solution, however, removing the interface regions may not be
considered.
Summary
We have here discussed some plausible reasons for why an evaluated
equilibrium salt concentration in a clay sample may be overestimated:
If samples are saturated directly with a saline solution. Better practice is to first saturate the sample with pure water (or a dilute solution) and then to equilibrate with respect to salt in a second stage.
If the external solution is not circulated. Diffusion may then occur over very long distances (depending on test design). The reasonable practice is to always circulate external solutions.
If interface excess is not handled. This is an issue even if saturation is done with pure water. The most convenient way to deal with this is to section off the first millimeters on both sides of the samples as quickly as possible after they are unloaded.
Footnotes
[1] One way to minimize this possible effect could be to
empty the filter before unloading the test cell. This may, however,
be difficult unless the filter itself is flushable. Also, you may
run into the problem of beginning to dry the sample.
[2] The only study I’m aware of that has
systematically investigated these types of concentration profiles is
Glaus et
al. (2011). They claim, if I understand correctly, that the
interface excess is not caused by swelling during
dismantling. Rather, they mean that the profile is the result of an
intrinsic density decrease that occurs in interface regions. Still,
they don’t discuss how swelling are supposed to be inhibited,
neither during dismantling, nor in order for the density
inhomogeneity to remain. Under any circumstance, the conclusions in
this blog post are not dependent on the cause for the presence of a
salt interface excess.
[3] In through-diffusion tests, the problem of the
interface excess is usually not that the equilibrium clay
concentration is systematically overestimated, since the detailed
concentration profile often is sampled in the final state. Instead,
the problem becomes how to separate the linear and non-linear parts
of the profile.
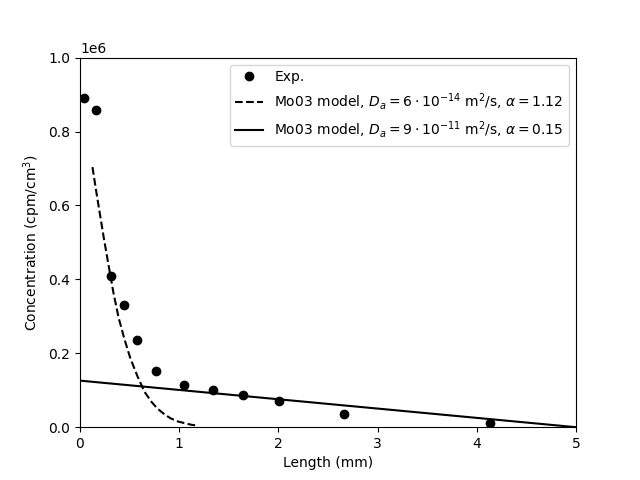
Mo03 performed both chloride and iodide through-diffusion tests on
“MX-80” bentonite, but here we focus on the chloride
results. However, since the only example in the paper of an outflux
evolution and corresponding concentration profile is for iodide, this
particular result will also be investigated. The tests were performed
at background concentrations of 0.01 M or 0.1 M NaClO4, and nominal
sample densities of 0.4, 0.8, 1.2, 1.6, and 1.8 g/cm3. We refer to a
single test by stating “nominal density/background concentration”,
e.g. a test performed at nominal density 1.6 and background
concentration 0.1 M is referred to as “1.6/0.1”.
Uncertainty of samples
The material used is discussed only briefly, and the only reference given for its properties is (Müller-Von Moos and Kahr, 1983). I don’t find any reason to believe that the “MX-80” batch used in this study actually is the one investigated in this reference, and have to assume the same type of uncertainty regarding the material as we did in the assessment of Muurinen et al (1988). I therefore refer to that blog post for a discussion on uncertainty in montmorillonite content, cation population, and soluble calcium minerals.
Density
The samples in Mo03 are cylindrical with radius 0.5 cm and length 0.5
cm, giving a volume of 0.39 cm3. This is quite small, and corresponds
e.g. only to about 4% of the sample size used in
Muurinen et al
(1988). With such a small volume, the samples are at the
limit for being considered as a homogeneous material, especially for
the lowest densities: the samples of density 0.4 g/cm3 contain 0.157 g
dry substance in total, while a single 1 mm3 accessory grain weighs
about 0.002 — 0.003 g.
Furthermore, as the samples are sectioned after termination, the
amount substance in each piece may be very small. This could cause
additional problems, e.g. enhancing the effect of drying. The
reported profile (1.6/0.1, iodide diffusion) has 10 sections in the
first 2 mm. As the total mass dry substance in this sample is 0.628 g,
these sections have about 0.025 g dry substance each (corresponding to
the mass of about ten 1 mm3 grains). For the lowest density, a similar
sectioning corresponds to slices of dry mass 0.006 g (the paper does
not give any information on how the low density samples were
sectioned).
Mo03 only report nominal densities for the samples, but from the above considerations it is clear that a substantial (but unknown) variation may be expected in densities and concentrations.
A common feature of many through-diffusion studies is that the sample
density appears to decrease in the first few millimeters near the
confining filters. We saw this effect in the profiles of
Muurinen et al (1988),
and it has been the topic of some
studies,
including Mo03. Here, we don’t consider any possible cause, but simply
note that the samples seem to show this feature quite generally (below
we discuss how Mo03 handle this). Since the samples of Mo03 are only
of length 5 mm, we may expect that the major part of them are affected
by this effect. Of course, this increases the uncertainty of the
actual density of the used samples.
Uncertainty of external solutions
Mo03 do not describe how the external solutions were prepared, other
than that they used high grade chemicals. We assume here that the
preparation did not introduce any significant uncertainty.
Since “MX-80” contains a substantial amount of divalent ions, connecting this material with (initially) pure sodium solutions inevitably initiates cation exchange processes. The extent of this exchange depends on details such as solution concentrations, reservoir volumes, number of solution replacements, time, etc…
Very little information is given on the volume of the external solution
reservoirs. It is only hinted that the outlet reservoir may be 25 ml,
and for the inlet reservoir the only information is
The volume of the inlet reservoir was sufficient to keep the
concentration nearly constant (within a few percent) throughout
the experiments.
Consequently, we do not have enough information to assess the exact ion population during the course of the tests. We can, however, simulate this process of “unintentional exchange” to get some appreciation for the amount of divalent ions still left in the sample, as we did in the assessment of Muurinen et al. (1988). Here are the results from calculating the exchange equilibrium between a sample initially containing 30% exchangeable charge in form of calcium (70% sodium), and external NaClO4 solutions of various concentrations and volumes
In these calculations we assume a sample of density 1.6 g/cm3 (except
when indicated), a volume of 0.39 cm3, a cation exchange capacity of
0.75 eq/kg, and a Ca/Na selectivity coefficient of 5.
These simulations make it clear that the tests performed at 0.01 M
most probably contain most of the divalent ions initially present in
the “MX-80” material: even with an external solution volume of 1000
ml, or with density 0.4 g/cm3, exchange is quite
limited. For the tests performed at 0.1 M we expect some exchange of
the divalent ions, but we really can’t tell to what extent, as the
exact value strongly depends on handling (solution volumes, if
solutions were replaced, etc.). That the exact ion population is
unknown, and that the divalent/monovalent ratio probably is different
for different samples, are obviously major problems of the study (the
same problems were identified
in Muurinen et al
(1988)).
Uncertainty of diffusion parameters
Diffusion model
Mo03 determine diffusion parameters by fitting a model to all
available data, i.e the outflux evolution and the concentration
profile across the sample at termination. The model is solved by a
numerical code (“ANADIFF”) that takes into account transport both in
clay samples and filters. The fitted parameters are an apparent
diffusivity, \(D_a\), and a so-called “capacity factor”,
\(\alpha\). \(\alpha\) is vaguely interpreted as being the combination of
a porosity factor \(\epsilon\), and a sorption distribution
coefficient \(K_d\), described as “a generic term devoid of mechanism”
It is claimed that for anions, \(K_d\) can be treated as negative, giving \(\alpha < \epsilon\). I have criticized this mixing of what actually are incompatible models in an earlier blog post. Strictly, this use of a “generic term devoid of mechanism” means that the evaluated \(\alpha\) should not be interpreted in any particular way. Nevertheless, the waythis study is referenced in otherpublications, \(\alpha\) is interpreted as an effective porosity. It should be noticed, however, that this study is performed with a background electrolyte of NaClO4. The only chloride (or iodide) present is therefore at trace level, and it cannot be excluded that a mechanism of true sorption influences the results (there are indications that this is the case in other studies).
For the present assessment we anyway assume that \(\alpha\) directly
quantifies the anion equilibrium between clay and the external
solution (i.e. equivalent to
the
incorrect way of
assuming that \(\alpha\) quantifies a volume accessible to
chloride). It should be kept in mind, though, that effects of anion
equilibrium and potential true sorption is not resolved by the
single parameter \(\alpha\).
where \(c\) is the concentration in the clay of the isotope under
consideration, and the diffusion coefficient is written \(D_p\) to
acknowledge that it is a pore diffusivity (when referring to models
and parameter evaluations in Mo03 we will use the notation
“\(D_a\)”). The boundary conditions are
Oddly, Mo03 model the system as if two independent diffusion processes are simultaneously active. They refer to these as the “fast” and the “slow” processes, and hypothesize that they relate to diffusion in interlayer water2 and “interparticle water”,3 respectively.
The “fast” process is the “ordinary” process that is assumed to reach steady state during the course of the test, and that is the focus of other through-diffusion studies. The “slow” process, on the other hand, is introduced to account for the frequent observation that measured tracer profiles are usually significantly non-linear near the interface to the source reservoir (discussed briefly above). I guess that the reason for this concentration variation is due to swelling when the sample is unloaded. But even if the reason is not fully clear, it can be directly ruled out that it is the effect of a second, independent, diffusion process — because this is not how diffusion works!
If anions move both in interlayers and “interparticle water”, they reasonably transfer back and forth between these domains, resulting in a single diffusion process (the diffusivity of such a process depends on the diffusivity of the individual domains and their geometrical configuration). To instead treat diffusion in each domain as independent means that these processes are assumed to occur without transfer between the domains, i.e. that the bentonite is supposed to contain isolated “interlayer pipes”, and “interparticle pipes”, that don’t interact. It should be obvious that this is not a reasonable assumption. Incidentally, this is how all multi-porous models assume diffusion to occur (while simultaneously assuming that the domains are in local equilibrium…).
We will thus focus on the “fast” process in this assessment, although we also use the information provided by the parameters for the “slow” process. Mo03 report the fitted values for \(D_a\) and \(\alpha\) in a table (and diagrams), and only show a comparison between model and measured data in a single case: for iodide diffusion at 0.1 M background concentration and density 1.6 g/cm3. To make any kind of assessment of the quality of these estimations we therefore have to focus on this experiment (the article states that these results are “typical high clay density data”).
Outflux
The first thing to note is that the modeled accumulated diffusive substance does not correspond to the analytical solution for the diffusion process. Here is a figure of the experimental data and the reported model (as presented in the article), that also include the solution to eqs. 1 and 2.
In fact, the model presented in Mo03 has an incorrect time dependency in the early stages. Here is a comparison between the presented model and analytical solutions in the transient stage
With the given boundary conditions, the solutions to the diffusion
equation inevitably has zero slope at \(t = 0\),4 reflecting
that it takes a finite amount of time for any substance to reach the
outflux boundary. The models presented in Mo03, on the other hand, has
a non-zero slope in this limit. I cannot understand the reason for
this (is it an underlying problem with the model, or just a graphical
error?), but it certainly puts all reported parameter values in doubt.
The preferred way to evaluate diffusion data is, in my opinion, to look
at the flux evolution rather than the evolution of the accumulated
amount of diffused substance. Converting the reported data to flux,
gives the following picture.5
From a flux evolution it is easier to establish the steady-state, as it reaches a constant. It furthermore gives a better understanding for how well constrained the model is by the data. As is seen from the figure, the model is not at all very well constrained, as the experimental data almost completely miss the transient stage. (And, again, it is seen that the model in the paper with \(D_a= 9\cdot 10^{-11}\) m/s2 does not correspond to the analytical solution.)
The short transient stage is a consequence of using thin samples (0.5 cm). Compared e.g. to Muurinen et al (1988), who used three times as long samples, the breakthrough time is here expected to be \(3^2 = 9\) times shorter. As Muurinen et al. (1988) evaluated breakthrough times in the range 1 — 9 days, we here expect very short times. Here are the breakthrough times for all chloride diffusion tests, evaluated from the reported diffusion coefficients (“fast” process) using the formula \(t_\mathrm{bt} = L^2/(6D_a)\).
Test
\(D_a\)
\(t_\mathrm{bt}\)
(m2/s)
(days)
0.4/0.01
\(8\cdot 10^{-10}\)
0.06
0.4/0.1
\(9\cdot 10^{-10}\)
0.05
0.4/0.1
\(8\cdot 10^{-10}\)
0.06
0.8/0.01
\(3.5\cdot 10^{-10}\)
0.14
0.8/0.1
\(3.5\cdot 10^{-10}\)
0.14
0.8/0.1
\(3.7\cdot 10^{-10}\)
0.13
1.2/0.01
\(1.4\cdot 10^{-10}\)
0.34
1.2/0.1
\(2.3\cdot 10^{-10}\)
0.21
1.2/0.1
\(2.0\cdot 10^{-10}\)
0.24
1.6/0.1
\(1.0\cdot 10^{-10}\)
0.48
1.8/0.01
\(2\cdot 10^{-11}\)
2.41
1.8/0.1
\(5\cdot 10^{-11}\)
0.96
1.8/0.1
\(5.5\cdot 10^{-11}\)
0.88
The breakthrough time is much shorter than a day in almost all tests! To sample the transient stage properly requires a sampling frequency higher than \(1/t_{bt}\). As seen from the provided example of a outflux evolution, this is not the case: The second measurement is done after about 1 day, while the breakthrough time is about 0.5 days (moreover, the first measurement appears as an outlier). We have no information on sampling frequency in the other tests, but note that to properly sample e.g. the tests at 0.8 g/cm3 requires measurements at least every third hour or so. For 0.4 g/cm3, the required sample frequency is once an hour! This design choice puts more doubt on the quality of the evaluated parameters.
Concentration profile
The measured concentration profile across the 1.6/0.1 iodide sample,
and corresponding model results are presented in Mo03 in a figure very
similar to this
Here the two models correspond to the “slow” and “fast” process discussed above (a division, remember, that don’t make sense). Zooming in on the “linear” part of the profile, we can compare the “fast” process with analytical solutions (eqs. 1 and 2)
The analytical solutions correspond directly to the outflux curves presented above. We note that the analytical solution with \(D_p = 9\cdot 10^{-11}\) m/s2 corresponds almost exactly to the model presented by Mo03. As this model basically has the same steady state flux and diffusion coefficient, we expect this similarity. It is, however, still a bit surprising, since the corresponding outflux curve of the model in Mo03 was seen to not correspond to the analytical solution. This continues to cast doubt on the model used for evaluating the parameters.
We furthermore note that the evolution of the activity of the source
reservoir is not reported. Once in the text is mentioned that the
“carrier concentration” is \(10^{-6}\) M, but since we don’t know how
much of this concentration corresponds to the radioactive isotope, we
can not directly compare with reported concentration profile across
the sample (whose concentration unit is counts per minute per cm3).
By extrapolating the above model curve with \(\alpha = 0.15\), we can
however deduce that the corresponding source activity for this
particular sample is \(C_0 = 1.26\cdot 10^5/0.15\) cpu/cm3
\(= 8.40\cdot 10^5\) cpu/cm3. But it is unsatisfying that we cannot
check this independently. Also, we can of course not assume that this
value of \(C_0\) is the same in any other of the tests (in particular
those involving chloride). We thus lack vital information (\(C_0\)) to
be able to make a full assessment of the model fitting.
It should furthermore be noticed that the experimental concentration
profile does not constrain the models very well. Indeed, the adopted
model (diffusivity \(9\cdot 10^{-11}\) m/s2) misses the two
rightmost concentration points (which corresponds to half the
sample!). A model that fits this part of the profile has a
considerable higher diffusivity, and a correspondingly lower
\(\alpha\) (note that the product \(D_p\cdot \alpha\) is constrained
by the steady-state flux, eq. 3).
More peculiarities of the modeling is found if looking at the “slow”
process (remember that this is not a real diffusion process!). Zooming
in on the interface part of the profile and comparing with analytical
solutions gives this picture
Here we note that an analytical solution coincides with the model presented in Mo03 with parameters \(D_a = 6\cdot 10^{-14}\) m2/s and \(\alpha = 1.12\) only if it is propagated for about 15 days! Given that no outflux measurements seem to have been performed after about 4 days (see above), I don’t now what to make of this. Was the test actually conducted for 15 days? If so, why is not more of the outflux measured/reported? (And why were the samples then designed to give a breakthrough time of only a few hours?)
Without knowledge of for how long the tests were conducted, the reported diffusion parameters becomes rather arbitrary, especially for the low density samples. For e.g. the samples of density 0.4 g/cm3, even the “slow” process has a diffusivity high enough to reach steady-state within a few days. Simulating the processes with the reported parameters gives the following profiles if evaluated after 1 and 4 days, respectively
The line denoted “total” is what should resemble the measured
(unreported) data. It should be clear from these plots that the
division of the profile into two separate parts is quite arbitrary. It
follows that the evaluated diffusion parameters for the process of
which we are interested (“fast”) has little value.
Summary and verdict
We have seen that the reported model fitting leaves a lot of unanswered questions: some of the model curves don’t correspond to the analytical solutions, information on evolution times and source concentrations is missing, and the modeled profiles are divided quite arbitrary into two separate contributions (which are not two independent diffusion process).
Moreover, the ion population (divalent vs. monovalent cations) of the samples are not known, but there are strong reasons to believe that the 0.01 M tests contain a significant amount of divalent ions, while the 0.1 M samples are partly converted to a more pure sodium state.
Also, the small size of the samples contributes to more uncertainty,
both in terms of density, but also for the flux evolution because the
breakthrough times becomes very short.
Based on all of these uncertainties, I mean that the results of Mo03
does not contribute to quantitative process understanding and my
decision is to not to use the study for e.g. validating models
of anion exclusion.
A confirmation of the uncertainty in this study is given by
considering the density dependence on the chloride equilibrium
concentrations for constant background concentration, evaluated from
the reported diffusion parameters (\(\alpha\) for the “fast” process).
If these results should be taken at face value, we have to accept a
very intricate density dependence: for 0.1 M background, the
equilibrium concentration is mainly constant between densities 0.3
g/cm3 and 0.7 g/cm3, and increases
between densities 1.0 g/cm3 and 1.45 g/cm3 (or,
at least, does not decrease). For 0.01 M background, the equilibrium
concentration instead falls quite dramatically between between
densities 0.3 g/cm3 and 0.7 g/cm3, and
thereafter displays only a minor density dependence.
To accept such dependencies, I require a considerably more rigorous experimental procedure and evaluation. In this case, I rather view the above plot as a confirmation of large uncertainties in parameter evaluation and sample properties.
[1] Strictly, \(c(0,t)\) relates to the concentration in the endpoint of the inlet filter. But we ignore filter resistance in this assessment, which is valid for the 1.6/0.1 sample. Moreover, the filter diffusivities are not reported in Mo03.
[2] Mo03 refer to interlayer pores as
“intralayer” pores, which may cause some confusion.
[3] Apparently, the authors assume an
underlying
stack view of the material.
[4] It may be
objected that the analytical solution do not include the filter
resistance. But note that filter resistance only will increase the
delay. Moreover, the transport capacity of the sample in this test
is so low that filters have no significant influence.
[5] The model by Mo03 looks noisy
because I have read off values of accumulated concentration from the
published graph. The “noise” occurs because the flux is evaluated
from the concentration data by the difference formula:
where \(t_i\) and \(t_{i+1}\) are the time coordinates for two consequitive data points, \(a(t)\) is the accumulated amount diffused substance at time \(t\), \(A\) is the cross sectional area of the sample, \(\bar{t}_i = (t_{i+1} + t_i)/2\) is the average time of the considered time interval, and \(\bar{j}\) denotes the average flux during this time interval.
We havediscussedvariousaspects of “anionexclusion” on this blog. This concept is often used to justify multi-porosity models of compacted bentonite, by reasoning that the exclusion mechanism makes parts of the pore space inaccessible to anions. But we have seen that this reasoning has no theoretical backup: studies making such assumptions usually turn out to refer to conventional electric double layer theory, described e.g. by the Poisson-Boltzmann equation. In the following, we refer to the notion of compartments inaccessible to anions as complete anion exclusion.
In fact, a single, physically reasonable concept underlies basically all descriptions of anion exclusion in the clay literature: charge separation. Although the required mathematics may differ for different systems — may it be using Donnan’s “classical equations”, or the Poisson-Boltzmann equation — the underlying mechanism is the same. In the following we refer to this type of description as traditional theory or Donnan theory. It is important to recognize that traditional theory is incompatible with complete anion exclusion: the Poisson-Boltzmann equation predicts anions everywhere.
In more recent years, however, a different meaning of the term “anion
exclusion” has sneaked into the literature. This seems to be related
to the dawn of molecular dynamics (MD) simulations of clays. In
particular, the study of Rotenberg et al. (2007) — which I think is the first published MD
simulation of montmorillonite interlayers in contact with an external
compartment — is frequently cited as demonstrating qualitatively
different results as compared with the traditional
models. E.g. Kosakowski and Berner (2013) write
Very often it is assumed that negatively charged ions are strongly
hindered to enter the interlayer space (Kosakowski et al., 2008;
Rotenberg et al., 2007), although other authors come to different
conclusions (Karnland et al., 2007). Note that we favor the former
view with our montmorillonite setup.
Although the terms “assumed” and “conclusions” seem misplaced, it
is clear that Kosakowski and Berner (2013) mean that the interlayer space is
essentially anion-free, rather than obeying ordinary Donnan
equilibrium (the approach used in
Karnland et
al. (2007)).
The interlayer space can be seen as an extreme case where the
diffuse layer vanishes leaving only the Stern layer of the adjacent
basal surfaces. For this reason, the interlayer space is often
considered to be completely free of anions (Tournassat and Appelo
2011), although this hypothesis is still controversial (Rotenberg et
al. 2007c; Birgersson and Karnland 2009).
Based upon [results from anion diffusion tests], anion-exclusion models have been formulated, which subdivide the water-filled pore space into interlayer, diffuse (or electric) double layer (DDL) and “free” water porosities (Wersin et al. 2004; Tournassat & Appelo 2011; Appelo 2013). In this formulation, anions are considered to reside in the “free” electrically neutral solution and in the DDL in the external (intergranular) pores, whereas the interlayer (intragranular) space is considered devoid of anions. Support for this model has been given by molecular dynamics simulations (Rotenberg et al. 2007), but this issue remains controversial (Birgersson & Karnland 2009)
The term “anion-exclusion” is here fully transformed to refer to complete exclusion, rather than to the traditional theory from which the term was coined. Note that the picture of bentonite given in this and the previous quotations is basically the contemporary mainstream view, which we discussed in a previous blog post. This description has not emerged from considering MD results that are allegedly in contradiction with traditional Donnan equilibrium theory. Rather, it has resulted from misusing the concept of exclusion-volume. The study of Rotenberg et al. (2007) (Rot07, in the following) supports the contemporary mainstream view only to the extent that it is at odds with the predictions of traditional theory. But is it? Let’s take a look at the relevant MD studies.
Rotenberg et al. (2007)
Rot07 is not primarily a study of the anion equilibrium, but considers more generally the transition of species between an external compartment2 and interlayer pores: water, cations (Na and Cs), and anions (Cl). The study only concerns interlayers with two monolayers of water, in the following referred to as a 2WL system. There is of course nothing wrong with exclusively studying the 2WL system, but this study alone cannot be used to support general model assumptions regarding interlayers (which anyway is commonplace, as we saw above). The meaning of the term “interlayer” in modern clay literature is quite confusing, but there is at least full consensus that it includes also states with three monolayers of water (3WL) (we’ll get back to those). Rot07 furthermore consider only a single external concentration, of 0.52 M.
Here is an illustration of the simulated system:
A cell (outlined with dashed lines) containing two montmorillonite
layers (yellow) and six chloride ions (green) is repeated infinitely
in all directions (the cell depth in the direction normal to the
picture is 20.72 Å). While only chloride ions are indicated in this
figure, also cations, water atoms, and montmorillonite atoms are
explicitly accounted for in the simulation.
Note that the study neither varies density (interlayer distance) nor
external concentration (number of chloride ions) — two variables
essential for studying anion equilibrium. I don’t mean this as direct
criticism, but it should be recognized when the study is used to
support assumptions regarding interlayers in other models.
What I do want to criticize, however, is that
Rot07 don’t
actually compare with Donnan theory. Instead, they seem to be under
the impression that traditional theory predicts complete exclusion in
their system. Consider this passage in the introduction
Due to the negative charge of clay layers, anions should be repelled
by the external surfaces, and excluded from the interlayers. On the
contrary, cations are attracted by the surfaces, and may exchange
with the natural interlayer counterions.
Here they associate two different terms with the anions: they are
repelled by the “external surfaces” and excluded from
“interlayers”.
I can only interpret this as meaning that anions are completely
excluded from interlayers, especially as the wording “on the
contrary” is used when describing cations.3
The study comprises both a “plain” MD simulation of the (presumed) equilibrium state, and separate calculations of free energy profiles. In the “plain” MD simulation, anions do not enter the interlayers, and the calculation of the free energy profile gives a barrier of ~9 kT for chloride to enter the interlayer.
These results motivate the authors to conclude that the “thermal fluctuations do not allow anions to overcome the free energy barrier corresponding to their entrance into the interlayer” and that “anions are excluded from the interlayer: the probability for an anion reaching the interface to enter into the interlayer is very small (of the order of e-9 ~ 10-4)”
It is important to keep in mind that the authors are under the
impression that this result and conclusion are in line with the
traditional description of anion
exclusion.3 When summarizing
their findings they write
All the results are in agreement with the common sense on ionic
exchange and anion exclusion.
and
The results confirm the generally admitted ideas of ionic exchange
and anion exclusion
The problem is that this “common sense” and these “generally
admitted ideas” are based on
misconceptions of traditional theory (I also think one should be
careful with using terms like these in scientific
writing). Consequently, the authors erroneously conclude that their
results confirm, rather than contrast, traditional theory. This is
opposite to how this study is referred to in later publications, as
was exemplified above.
The anion exclusion predicted from Donnan theory for the system in
Rot07 is estimated
as follows. The adopted montmorillonite unit cell
(Na0.75Si8Al3.25Mg0.75O20OH4)
has
structural charge 0.75e, and lateral dimensions 8.97 Å × 5.18
Å. With an interlayer width of 6.1 Å we thus have for the
concentration of interlayer charge
where \(N_A\) is the
Avogadro
constant. Using this value for \(c_{IL}\) in the expression for
internal anion concentration in an ideal
1:1 Donnan system,
This should be the anion interlayer concentration expected from “generally admitted ideas”, and Rot07 should have concluded that their results differ by a factor ~1000 (or more) from traditional theory. This is not to say that the calculations are incorrect (more on that later), but it certainly puts the results in a different light. A discrepancy of this magnitude should reasonably be of interest to investigate further.
Hsiao and Hedström (2015)
Considerably more detailed MD simulations of the 2WL system are
provided by Hsiao and
Hedström (2015) (Hsi15, hereafter). In contrast to
Rot07,
Hsi15 specifically
focus on the anion equilibrium, and they explicitly compare with both
conventional Donnan theory, and the results of
Rot07. In these
simulations, chloride actually populates the interlayer.
Hsi15 also analyze the convergence behavior, by varying system size and simulation time. This analysis makes it clear both that most of the simulations presented in the paper are properly converged, and that the simulation of Rot07 is not. With external concentration 1.67 M, Hsi15 demonstrate that, during intervals of 20 ns, the interlayer concentration fluctuates between basically zero and 0.13 M (converged value: 0.04 M), in a system with similar size as that of Rot07. Given that the total simulation time of the earlier study is 20 ns, and that it also adopts a considerably lower external concentration, its result of zero chloride concentration in the interlayer is no surprise.
The converged interlayer concentrations in
Hsi15 look like
this in the direction normal to the basal surfaces (simulation time:
150 ns, layer size: 8 × 4 unit cells, external concentration:
1.67 M)
Note that the simulation contains two interlayer pores (indicated by the dotted lines; cf. the illustration of the simulated system) and that sodium and chloride populate the same central layer, sandwiched by the two water layers (not shown). The nearly identical chloride profiles is a strong confirmation that the simulation is converged.
The chloride interlayer concentrations evaluated in Hsi15 deviate strongly from the predictions of the ideal Donnan formula. With \(c_{IL}\) = 4.23 M (as reported in the article) and \(c^\mathrm{ext}\) = 1.67 M, eq. 1 gives \(c^\mathrm{int}\) = 0.580 M, while the MD results are in the range 0.033 M — 0.045 M, i.e. more than a factor 10 lower (but not a factor 1000).
Hsi15 also calculate
the free energy profiles along the coordinate connecting the external
compartment and the interlayer, similar to the technique utilized by
Rot07 (as far as I
understand). For the external concentration of 1.67 M they evaluate a
free energy barrier of ~3.84 kT, which corresponds to an
interlayer concentration of 0.036 M, and is in good agreement with the
directly evaluated concentrations.
Note that Hsi15 —
in contrast to Rot07 — conclude significant deviation between the MD results of
the 2WL system and ideal traditional theory. Continuing their
investigation (again, in contrast to
Rot07),
Hsi15 found that the
contribution from ion hydration to the free energy barrier basically
make up for the entire discrepancy with the ideal Donnan formula.
Moreover, even though the ideal Donnan formula strongly overestimates
the actual values obtained from MD, it still shows the correct
dependency on external concentration: when the external
concentration is lowered to 0.55 M, the evaluated free energy barrier
increases to ~5.16 kT, which corresponds to a reduction of the
internal concentration by about a factor of 10. This is in
agreement with Donnan theory, which gives for the expected
reduction (0.55/1.67)2 ≈ 0.11.
From the results of Hsi15 (and Rot07,
for that matter), a relatively clear picture emerges: MD simulated 2WL
systems function as Donnan systems. Anions are not completely
excluded, and the dependency on external concentration is in line with
what we expect from
a varying Donnan potential across the interface between interlayer
and external compartment
(Hsi15 even comment
on observing the space-charge region!).
The simulated 2WL system is, however, strongly non-ideal, as a consequence of the ions not being optimally hydrated. Hsi15 remark that the simulations probably overestimate this energy cost, e.g. because atoms are treated as non-polarizable. This warning should certainly be seriously considered before using the results of MD simulated 2WL systems to motivate multi-porosity in compacted bentonite. But, concerning assumptions of complete anion exclusion in interlayers, another system must obviously also be considered: 3WL.
Hedström and Karnland (2012)
MD simulations of anion equilibrium in the 3WL system are presented in
Hedström and
Karnland (2012) (Hed12, in the following).
Hed12 consider
three different external concentrations, by including either 12, 6, or
4 pairs of excess ions (Cl– + Na+). This study
also varies the way the interlayer charge is distributed, by either
locating unit charges on specific magnesium atoms in the
montmorillonite structure, or by evenly reducing the charge by a minor
amount on all the octahedrally coordinated atoms.
Here are the resulting ion concentration profiles across the
interlayer, for the simulation containing 12 chloride ions, and evenly
distributed interlayer charge (simulation time: 20 ns, layer size:
4 × 4 unit cells)
Chloride mainly resides in the middle of the interlayer also in the 3WL system, but is now separated from sodium, which forms two off-center main layers. The dotted lines indicate the extension of the interlayer.
The main objectives of this study are to simply establish that anions in MD equilibrium simulations do populate interlayers, and to discuss the influence of unavoidable finite-size effects (6 and 12 are, after all, quite far from Avogadro’s number). In doing so, Hed12 demonstrate that the system obeys the principles of Donnan equilibrium, and behaves approximately in accordance with the ideal Donnan formula (eq. 1). The authors acknowledge, however, that full quantitative comparison with Donnan theory would require better convergence of the simulations (the convergence analysis was further developed in Hsi15). If we anyway make such a comparison, it looks like this
#Cl TOT
Layer charge
#Cl IL
\(c^\mathrm{ext}\)
\(c^\mathrm{int}\) (Donnan)
\(c^\mathrm{int}\) (MD)
12
distr.
1.8
1.45
0.62
0.42 (67%)
12
loc.
1.4
1.50
0.66
0.32 (49%)
6
distr.
0.6
0.77
0.20
0.14 (70%)
6
loc.
1.3
0.67
0.15
0.30 (197%)
4
distr.
0.2
0.54
0.10
0.05 (46%)
4
loc.
0.18
0.54
0.10
0.04 (41%)
The first column lists the total number of chloride ions in the simulations, and the second indicates if the layer charge was distributed on all octahedrally coordinated atoms (“distr.”) or localized on specific atoms (“loc.”) The third column lists the average number of chloride ions found in the interlayer in each simulation. \(c^\mathrm{ext}\) denotes the corresponding average molar concentration in the external compartment. The last two columns lists the corresponding average interlayer concentration as evaluated either from the Donnan formula (eq. 1 with \(c_{IL}\) = 2.77 M, and the listed \(c^\mathrm{ext}\)), or from the simulation itself.
The simulated results are indeed within about a factor of 2 from the predictions of ideal Donnan theory, but they also show a certain variation in systems with the same number of total chloride ions,4 indicating incomplete convergence (compare with the fully converged result of Hsi15). It is also clear from the analysis in Hed12 and Hsi15 that the simulations with the highest number och chloride ions (12) are closer to being fully converged.5 Let’s therefore use the result of those simulations to compare with experimental data.
Comparison with experiments
In an earlier blog post, we looked at the available experimental data on chloride equilibrium concentrations in Na-dominated bentonite. Adding the high concentration chloride equilibrium results from Hed12 and Hsi15 to this data (in terms of \(c^\mathrm{int}/c^\mathrm{ext}\)), gives the following picture6 (the 3WL system corresponds to pure montmorillonite of density ~1300 kg/m3, and the 2WL system corresponds to ~1600 kg/m3, as also verified experimentally).
The x-axis shows montmorillonite effective dry density, and applied external concentrations for each data series are color coded, but also listed in the legend. Note that this plot contains mainly all available information for drawing conclusions regarding anion exclusion in interlayers.7 To me, the conclusions that can be drawn are to a large extent opposite to those that have been drawn:
The amount chloride in the simulated 3WL system corresponds roughly to measured values. Consequently, MD simulations do not support models that completely exclude anions from interlayers.
The 3WL results instead suggest that interlayers contain the main contribution of chloride. Interlayers must consequently be handled no matter how many additional pore structures a model contains.
For systems corresponding to 2WL interlayers, there is a choice: Either,
assume that the discrepancy between simulations and measurements indicates the existence of an additional pore structure, where the majority of chloride resides, or
assume that presently available MD simulations of 2WL systems overestimate “anion” exclusion.8
Tournassat et al. (2016) (Tou16, in the following) present more MD simulations of interlayer pores in contact with an external compartment, with a fixed amount of excess ions, at three different interlayer distances: 2WL (external concentration ~0.5 M), 3WL (~0.4 M), and 5WL (~0.3 M).
In the 2WL simulations, no anions enter the interlayers. Tou16 do not reflect on the possibility that 2WL simulations may overestimate exclusion, as suggested by Hsi159, but instead use this result to argue that anions are basically completely excluded from 2WL interlayers. They even imply that the result of Rot07 is more adequate than that of Hsi15
In the case of the 2WL hydrate, no Cl– ion entered the
interlayer space during the course of the simulation, in agreement
with the modeling results of Rotenberg et al. (2007b), but in
disagreement with those of Hsiao and Hedström (2015).
But, as discussed, there is no real “disagreement” between the
results of Hsi15 and
Rot07. To refute
the conclusions of Hsi15, Tou16 are
required to demonstrate well converged results, and analyze what is
supposedly wrong with the simulations of
Hsi15. It is,
furthermore, glaringly obvious that most of the anion equilibrium
results in Tou16
are not converged.
Regarding convergence, the only “analysis” provided is the following
passage
The simulations were carried out at the same temperature (350 K) as the simulations of Hsiao and Hedström (2015) and with similar simulation times (50 ns vs. 100-200 ns) and volumes (27 × 104 Å3vs. 15 × 104 Å3), thus ensuring roughly equally reliable output statistics. The fact that Cl– ions did not enter the interlayer space cannot, therefore, be attributed to a lack of convergence in the present simulation, as Hsiao and Hedström have postulated to explain the difference between their results and those of Rotenberg et al. (2007b).
I mean that this is not a suitable procedure in a scientific
publication — the authors should of course demonstrate convergence of
the simulations actually performed! (Especially after
Hsi15 have provided
methods for such an analysis.10)
Anyhow, Tou16 completely miss that Hsi15 demonstrate convergence in simulations with external concentration 1.67 M; for the system relevant here (0.55 M), Hsi15 explicitly write that the same level of convergence requires a 10-fold increase of the simulation time (because the interlayer concentration decreases approximately by a factor of 10, as predicted by — Donnan theory). Thus, the simulation time of Tou16 (53 ns) should be compared with 2000 ns, i.e. it is only a few percent of the time required for proper convergence.
Further confirmation that the simulations in
Tou16 are not
converged is given by the data for the systems where chloride
has entered the interlayers. The ion concentration profiles for
the 3WL simulation look like this
The extension of the interlayers is indicated by the dotted lines. Each interlayer was given slightly different (average) surface charge density, which is denoted in the figure. One of the conspicuous features of this plot is the huge difference in chloride content between different interlayers: the concentration in the mid-pore (0.035 M) is more than three times that in left pore (0.010 M). This clearly demonstrates that the simulation is not converged (cf. the converged chloride result of Hsi15). Note further that the larger amount of chloride is located in the interlayer with the highest surface charge, and the least amount is located in the interlayer with the smallest surface charge.11 I think it is a bit embarrassing for Clays and Clay Minerals to have used this plot for the cover page.
As the simulation times (53 ns vs. 40 ns), as well as the external concentrations (~0.5 M vs. ~0.4 M), are similar in the 2WL and and 3WL simulations, it follows from the fact that the 3WL system is not converged, that neither is the 2WL system. In fact, the 2WL system is much less converged, given the considerably lower expected interlayer concentration. This conclusion is fully in line with the above consideration of convergence times in Hsi15.
For chloride in the 3WL (and 5WL) system, Tou16 conclude that “reasonable quantitative agreement was found” between MD and traditional theory, without the slightest mentioning of what that implies.12 I find this even more troublesome than the lack of convergence. If the authors mean that MD simulations reveal the true nature of anion equilibrium (as they do when discussing 2WL), they here pull the rug out from under the entire mainstream bentonite view! With the 3WL system containing a main contribution, interlayers can of course not be modeled as anion-free, as we discussed above. Yet, not a word is said about this in Tou16.
In this blog post I have tried to show that available MD simulations do not, in any reasonable sense, support the assumption that anions are completely excluded from interlayers. Frankly, I see this way of referencing MD studies mainly as an “afterthought”, in attempts to justify the misuse of the exclusion-volume concept. In this light, I am not surprised that Hed12 and Hsi15 have not gained reasonable attention, while Tou16 nowadays can be found referenced to support claims that anions do not have access to “interlayers”.13
Footnotes
[1] I should definitely
discuss the “Stern layer” in a future blog post.
[2] The view of bentonite (“clay”) in Rotenberg et al. (2007) is strongly rooted in a “stack” concept. What I refer to as an “external compartment” in their simulation, they actually conceive of as a part of the bentonite structure, calling it a “micropore”.
[3] That
Rotenberg et
al. (2007) expresses this view of anion exclusion puzzles me
somewhat, since several of the same authors published a study just a
few years later where Donnan theory was explored in similar systems:
Jardat et al. (2009).
[4] Since the number of chloride ions found in the
interlayer is not correlated with how layer charge is distributed,
we can conclude that the latter parameter is not important for the
process.
[5] The small difference in the two
simulations with 4 chloride ions is thus a coincidence.
[6] I am in the process of
assessing the experimental data, and hope to be able to better
sort out which of these data series are more relevant. So far I have
only looked at — and discarded —
the
study by Muurinen et al. (1988). This study is therefore removed
from the plot.
[7] There are of course severalotherresults that indirectly demonstrate the presence of anions in interlayers. Anyway, I think that the bentonite research community, by now, should have managed to produce better concentration data than this (both simulated and measured).
[8] As the cation (sodium) may give a major contribution to the hydration energy barrier (this is not resolved in Hsiao and Hedström (2015)), it may be inappropriate to refer to this part as “anion” exclusion (remember that it is salt that is excluded from bentonite). It may be noted that sodium actually appear to have a hydration barrier in e.g. the Na/Cs exchange process, which has been explored both experimentally and in MD simulations.
[9] Tournassat et al. (2016) even refer to Hsiao and Hedström (2015) as presenting a “hypothesis” that “differences in solvation energy play an important role in inhibiting the entry of Cl– in the interlayer space”, rather than addressing their expressed concern that the hydration energy cost may be overestimated.
[11] As the interlayers have different surface charge, they are not expected to have identical chloride content. But the chloride content should reasonably decrease with increasing surface charge, and the difference between interlayers should be relatively small.
[12] Here we have to disregard that the “agreement” is not quantitative. It is not even qualitative: the highest chloride content was recorded in the interlayer pore with highest charge, in both the 3WL and the 5WL system.
where \(\phi\) is the porosity of the sample, \(D_c\) is the macroscopic
pore diffusivity of the presumed interlayer domain, and \(\Xi\) is the
ion equilibrium coefficient. \(\Xi\) quantifies the ratio between
internal and external concentrations of the ion under consideration,
when the two compartments are in equilibrium.
where \(\epsilon_\mathrm{eff}\) is the porosity of a presumed bulk water
domain where anions are assumed to reside exclusively, and \(D_p\) is
the corresponding pore diffusivity of this bulk water domain.
We have
discussed earlier
how the homogeneous mixture and the effective porosity models can be
equally well fitted to a specific set of anion through-diffusion
data. The parameter “translation” is simply
\(\phi\cdot \Xi \leftrightarrow \epsilon_\mathrm{eff}\) and
\(D_c \leftrightarrow D_p\). It may appear from this equivalency that
diffusion data alone cannot be used to discriminate between the two
models.
But note that the interpretation of how \(D_e\) varies with background
concentration is very different in the two models.
In the homogeneous mixture model, \(D_c\) is not expected to vary
with background concentration to any greater extent, because the
diffusing domain remains essentially the same. \(D_e\) varies in this
model primarily because \(\Xi\) varies with background concentration, as
a consequence of an altered
Donnan potential.
In the effective porosity model, \(D_p\) is expected to vary,
because the volume of the bulk water domain, and hence the entire
domain configuration (the “microstructure”), is postulated to vary
with background concentration. \(D_e\) thus varies in this model both
because \(D_p\) and \(\epsilon_\mathrm{eff}\) varies.
A simple way of taking into account a varying domain configuration (as in the effective porosity model) is to assume that \(D_p\) is proportional to \(\epsilon_\mathrm{eff}\) raised to some power \(n – 1\), where \(n > 1\). Eq. 2 can then be written
where \(D_0\) is the tracer diffusivity in pure bulk water. Eq. 3 is in the bentonite literature often referred to as “Archie’s law”, in analogy with a similar evaluation in more conventional porous systems. Note that with \(D_0\) appearing in eq. 3, this expression has the correct asymptotic behavior: in the limit of unit porosity, the effective diffusivity reduces to that of a pure bulk water domain.
Eq. 3 shows that \(D_e\) in the effective porosity model is expected to depend non-linearly on background concentration for constant sample density. In contrast, since \(D_c\) is not expected to vary significantly with background concentration, we expect a linear dependence of \(D_e\) in the homogeneous mixture model. Keeping in mind the parameter “translation” \(\phi\cdot\Xi \leftrightarrow \epsilon_\mathrm{eff}\), the prediction of the homogeneous mixture model (eq. 1) can be expressed1
We have thus managed to establish a testable difference between the effective porosity and the homogeneous mixture model (eqs. 3 and 4). This is is great! Making this comparison gives us a chance to increase our process understanding.
Comparison with experiment
Van Loon et al. (2007)
It turns out that the chloride diffusion measurements performed by Van Loon et al. (2007) are accurate enough to resolve whether \(D_e\) depends on “\(\epsilon_\mathrm{eff}\)” according to eqs. 3 or 4. As will be seen below, this data shows that \(D_e\) varies in accordance with the homogeneous mixture model (eq. 4). But, since Van Loon et al. (2007) themselves conclude that \(D_e\) obeys Archie’s law, and hence complies with the effective porosity model, it may be appropriate to begin with some background information.
Van Loon et al. (2007) report three different series of diffusion tests, performed on bentonite samples of density 1300, 1600, and 1900 kg/m3, respectively. For each density, tests were performed at five different NaCl background concentrations: 0.01 M, 0.05 M, 0.1 M, 0.4 M, and 1.0 M. The tests were evaluated by fitting the effective porosity model, giving the effective diffusion coefficient \(D_e\) and corresponding “effective porosity” \(\epsilon_\mathrm{eff}\) (it is worth repeating that the latter parameter equally well can be interpreted in terms of an ion equilibrium coefficient).
Van Loon et al. (2007) conclude that their data complies with eq. 3, with \(n = 1.9\), and provide a figure very similar to this one
Here are compared evaluated values of effective diffusivity and “effective porosity” in various tests. The test series conducted by Van Loon et al. (2007) themselves are labeled with the corresponding sample density, and the literature data is from García-Gutiérrez et al. (2006)2 (“Garcia 2006”) and the PhD thesis of A. Muurinen (“Muurinen 1994”). Also plotted is Archie’s law with \(n\) =1.9. The resemblance between data and model may seem convincing, but let’s take a further look.
Rather than lumping together a whole bunch of data sets, let’s focus on the three test series from Van Loon et al. (2007) themselves, as these have been conducted with constant density, while only varying background concentration. This data is thus ideal for the comparison we are interested in (we’ll get back to commenting on the other studies).
It may also be noted that the published plot contains more data points (for these specific test series) than are reported in the rest of the article. Let’s therefore instead plot only the tabulated data.3 The result looks like this
Here we have also added the predictions from the homogeneous mixture model (eq. 4), where \(D_c\) has been fitted to each series of constant density.
The impression of this plot is quite different from the previous one: it should be clear that the data of Van Loon et al. (2007) agrees fairly well with the homogeneous mixture model, rather than obeying Archie’s law. Consequently, in contrast to what is stated in it, this study refutes the effective porosity model.
The way the data is plotted in the article is reminiscent of Simpson’s paradox: mixing different types of dependencies of \(D_e\) gives the illusion of a model dependence that really isn’t there. Reasonably, this incorrect inference is reinforced by using a log-log diagram (I have warned about log-log plots earlier). With linear axes, the plots give the following impression
This and the previous figure show that \(D_e\) depends approximately linearly on “\(\epsilon_\mathrm{eff}\)”, with a slope dependent on sample density. With this insight, we may go back and comment on the other data points in the original diagram.
García-Gutiérrez et al. (2006) and Muurinen et al. (1988)
The tests by García-Gutiérrez et al. (2006) don’t vary the background concentration (it is not fully clear what the background concentration even is4), and each data point corresponds to a different density. This data therefore does not provide a test for discriminating between the models here discussed.
I have had no access to Muurinen (1994), but by examining the data, it is clear that it originates from Muurinen et al. (1988), which was assessed in detail in a previous blog post. This study provides two estimations of “\(\epsilon_\mathrm{eff}\)”, based on either breakthrough time or on the actual measurement of the final state concentration profile. In the above figure is plotted the average of these two estimations.5
One of the test series in Muurinen et al. (1988) considers variation of density while keeping background concentration fixed, and does not provide a test for the models here discussed. The data for the other two test series is re-plotted here, with linear axis scales, and with both estimations for “\(\epsilon_\mathrm{eff}\)”, rather than the average6
As discussed in the assessment of this study, I judge this data to be too uncertain to provide any qualitative support for hypothesis testing. I think this plot confirms this judgment.
Glaus et al. (2010)
The measurements by Van Loon et al. (2007) are enough to convince me that the dependence of \(D_e\) for chloride on background concentration is furtherevidence for that a homogeneous view of compacted bentonite is principally correct. However, after the publication of this study, the same authors (partly) published more data on chloride equilibrium, in pure Na-montmorillonite and “Na-illite”,7 in Glaus et al. (2010).
This data certainly shows a non-linear relation between \(D_e\) and “\(\epsilon_\mathrm{eff}\)” for Na-montmorillonite, and Glaus et al. (2010) continue with an interpretation using “Archie’s law”. Here I write “Archie’s law” with quotation marks, because they managed to fit the expression to data only by also varying the prefactor. The expression called “Archie’s law” in Glaus et al. (2010) is
where \(A\) is now a fitting parameter. With \(A\) deviating from \(D_0\), this expression no longer has the correct asymptotic behavior as expected when interpreting \(\epsilon_\mathrm{eff}\) as quantifying a bulk water domain (see eq. 3). Nevertheless, Glaus et al. (2010) fit this expression to their measurements, and the results look like this (with linear axes)
Here is also plotted the prediction of the homogeneous mixture model
(eq. 4). For the montmorillonite data, the dependence is
clearly non-linear, while for the “Na-illite” I would say that the
jury is still out.
Although the data for montmorillonite in
Glaus et al. (2010)
is
non-linear, there are several strong arguments for why this is not an
indication that the effective porosity model is correct:
Remember that this result is not a confirmation of the measurements in Van Loon et al. (2007). As demonstrated above, those measurements complies with the homogeneous mixture model. But even if accepting the conclusion made in that publication (that Archie’s law is valid), the Glaus et al. (2010) results do not obey Archie’s law (but “Archie’s law”).
The four data points correspond to background concentrations of 0.1 M, 0.5 M, 1.0 M, and 2.0 M. If “\(\epsilon_\mathrm{eff}\)” represented the volume of a bulk water phase, it is expected that this value should level off, e.g. as the Debye screening length becomes small (Van Loon et al. (2007) argue for this). Here “\(\epsilon_\mathrm{eff}\)” is seen to grow significantly, also in the transition between 1.0 M and 2.0 M background concentration.
These are Na-montmorillonite samples of dry density 1.9 g/cm3. With an “effective porosity” of 0.067 (the 2.0 M value), we have to accept more than 20% “free water” in these very dense systems! This is not even accepted by otherproponents of bulk water in compacted bentonite.
Furthermore, these tests were performed with a background of \(\mathrm{NaClO_4}\), in contrast to Van Loon et al. (2007), who used chloride also for the background. The only chloride around is thus at trace level, and I put my bet on that the observed non-linearity stems from sorption of chloride on some system component.
Insight from closed-cell tests
Note that the issue whether or not \(D_e\) varies linearly with
“\(\epsilon_\mathrm{eff}\)” at constant sample density is equivalent
to whether or not \(D_p\) (or \(D_c\)) depends on background
concentration. This is similar to how presumed concentration
dependencies of the pore diffusivity for simple cations
(“apparent”
diffusivities) have been used to argue for multi-porosity in compacted
bentonite. For cations,
a closer look shows that no such dependency is found in the
literature.
For anions, it is a bit frustrating that the literature data is not
accurate or relevant enough to fully settle this issue (the data of
Van Loon et al. (2007)
is, in my opinion, the best available).
However, to discard the conceptual view underlying the effective porosity model, we can simply use results from closed-cell diffusion studies. In Na-montmorillonite equilibrated with deionized water, Kozaki et al. (1998) measured a diffusivity of \(1.8\cdot 10^{-11}\) m2/s at dry density 1.8 g/cm3.8 If the effective porosity hypothesis was true, we’d expect a minimal value for the diffusion coefficient9 in this system, since \(\epsilon_\mathrm{eff}\) approaches zero in the limit of vanishing ionic strength. Instead, this value is comparable to what we can evaluate from e.g. Glaus et al. (2010) at 1.9 cm3/g, and 2.0 M background electrolyte: \(D_e/\epsilon_\mathrm{eff} = 7.2\cdot 10^{-13}/0.067\) m2/s = \(1.1\cdot 10^{-11}\) m2/s.
That chloride diffuses just fine in dense montmorillonite equilibrated with pure water is really the only argument needed to debunk the effective porosity hypothesis.
Footnotes
[1] Note that \(\epsilon_\mathrm{eff}\) is not a parameter in the homogeneous mixture model, so eq. 4 looks a bit odd. But it expresses \(D_e\) if \(\phi\cdot \Xi\) is interpreted as an effective porosity.
[3] This choice is not critical for the conclusions made in this blog post, but it seems appropriate to only include the data points that are fully described and reported in the article.
[4] García-Gutiérrez et al. (2004) (which is the study compiled in García-Gutiérrez et al. (2006)) state that the samples were saturated with deionized water, and that the electric conductivity in the external solution were in the range 1 — 3 mS/cm.
[5] The data point labeled with a “?” seems to have been obtained by making this average on the numbers 0.5 and 0.08, rather than the correctly reported values 0.05 and 0.08 (for the test at nominal density 1.8 g/cm3 and background concentration 1.0 M).
[6] Admittedly, also the data we have plotted from the original tests in Van Loon et al. (2007) represents averages of several estimations of “\(\epsilon_\mathrm{eff}\)”. We will get back to the quality of this data in a future blog post when assessing this study in detail, but it is quite clear that the estimation based on the direct measurement of stable chloride is the more robust (it is independent of transport aspects). Using these values for “\(\epsilon_\mathrm{eff}\)”, the corresponding plot looks like this
[7] To my mind, it is a misnomer to describe something as illite in sodium form. Although “illite” seems to be a bit vaguely defined, it is clear that it is supposed to only contain potassium as counter-ion (and that these ions are non-exchangeable; the basal spacing is \(\sim\)10 Å independent of water conditions). The material used in Glaus et al. (2010) (and severalotherstudies) has a stated cation exchange capacity of 0.22 eq/kg, which in a sense is comparable to the montmorillonite material (a factor 1/4). Shouldn’t it be more appropriate to call this material e.g. “mixed-layer”?
[8] This value is the average from two tests performed at 25 °C. The data from this study is better compiled in Kozaki et al. (2001).
[9] Here we refer of course to the empirically defined diffusion coefficient, which I have named \(D_\mathrm{macr.}\) in earlier posts. This quantity is model independent, but it is clear that it should be be associated with the pore diffusivities in the two models here discussed (i.e. with \(D_c\) in the homogeneous mixture model, and with \(D_p\) in the effective porosity model).
Mu88 performed both chloride and uranium through-diffusion tests on “MX-80” bentonite, as well as sorption tests. Here we focus solely on the chloride diffusion. We also disregard one diffusion test series that does not vary external concentration (it was conducted with an unspecified “artificial groundwater” and varied sample density).
Left are two test series performed with nominal sample densities 1.2 g/cm3 and 1.8 g/cm3, respectively. For each of these densities, chloride through-diffusion tests were performed with external NaCl concentrations of 0.01 M, 0.1 M, and 1.0 M, respectively. The samples were cylindrical with a diameter of 3.0 cm, and a length of 1.5 cm, giving a volume of 10.6 cm3. To refer to a specific test or sample, we use the nomenclature “nominal density/external concentration”, e.g. the test performed at nominal density 1.2 g/cm3 and external solution 0.1 M is referred to as “1.2/0.1”.
Uncertainty of bentonite samples
“MX-80” is not the name of some specific standardized material, but simply a product name.2 It is quite peculiar that that “MX-80” nevertheless is a de facto standard in the research field for clay buffers in radwaste repositories. But, being a de facto standard, several batches of bentonite with this name have been investigated and reported throughout the years. We consequently have some appreciation for its constitution, and the associated variation.
In Mu88, the material used is only mentioned by name, and it is only
mentioned once (in the abstract!). We therefore can’t tell which of
the studies that is more appropriate to refer to. Instead, let’s take
a look at how “MX-80” has been reported generally.
*) These values were derived from summing the exchangeable ions, and are probably overestimations.
Montmorillonite content
Reported montmorillonite content varies in the range 75 — 85%. For the present context, this primarily gives an uncertainty in adopted effective montmorillonite dry density, which, in turn, is important for making relevant comparison between bentonite materials with different montmorillonite content. For the “MX-80” used in Mu88 we here assume a montmorillonite content of 80%. In the table below is listed the corresponding effective montmorillonite densities when varying the montmorillonite content in the range \(x =\) 0.75 — 0.85, for the two nominal dry densities.
Dry density
EMDD (\(x\)=0.75)
EMDD (\(x\)=0.80)
EMDD (\(x\)=0.85)
(g/cm3)
(g/cm3)
(g/cm3)
(g/cm3)
1.2
1.01
1.05
1.09
1.8
1.61
1.66
1.70
The uncertainty in montmorillonite content thus translates to an
uncertainty in effective montmorillonite dry density on the order of
0.1 g/cm3.
Cation population
While reported values of the cation exchange capacity of “MX-80” are relatively constant, of around 0.75 eq/kg,4 the reported fraction of sodium ions is seen to vary, in the range 70 — 85 %. The remaining population is mainly di-valent rare-earth metal ions (calcium and magnesium). This does not only mean that different studies on “MX-80” may give results for quite different types of systems, as the mono- to di-valent ion ratio may vary, but also that samples within the study may represent quite different systems. We examine this uncertainty below, when discussing the external solutions.
Soluble calcium minerals
The uncertainty of how much divalent cations are available is in fact larger than just discussed. “MX-80” is reported to contain a certain amount of soluble calcium minerals, in particular gypsum. These provide additional sources for divalent ions, which certainly will be involved in the chemical equilibration as the samples are water saturated. Reported values of gypsum content in “MX-80” are on the order of 1%. With a molar mass of 0.172 kg/mol, this contributes to the calcium content by \(2\cdot 0.01/0.172\) eq/kg \(\approx 0.12\) eq/kg, or about 16% of the cation exchange capacity.
Sample density
The samples in Mu88 that we focus on have nominal dry density of 1.2
and 1.8 g/cm3. The paper also reports measured porosities on each
individual sample, listed in the below table together with
corresponding values of dry density5
Test
\(\phi\)
\(\rho_d\)
(-)
(g/cm3)
1.2/0.01
0.54
1.27
1.2/0.1
0.52
1.32
1.2/1.0
0.49
1.40
1.8/0.01
0.37
1.73
1.8/0.1
0.31
1.89
1.8/1.0
0.34
1.81
We note a substantial variation in measured density for samples with the same nominal density: for the 1.2 g/cm3 samples, the standard deviation is 0.06 g/cm3, and for the 1.8 g/cm3 samples it is 0.07 g/cm3. Moreover, while the mean value for the 1.8 g/cm3 samples is close to the nominal value (1.81 g/cm3), that for the 1.2 g/cm3 samples is substantially higher (1.33 g/cm3).
It is impossible to know from the information provided in Mu88 if this
uncertainty is intrinsic to the procedure of preparing the samples, or
if it is more related to the procedure of measuring the density at
test termination.6
Uncertainty of external solutions
Mu88 do not describe how the external solutions were prepared. We
assume here, however, that preparing pure NaCl solutions gives no
significant uncertainty.
Further, the paper contains no information on how the samples were water saturated, nor on the external solution volumes. Since samples with an appreciable amount of di-valent cations are contacted with pure sodium solutions, it is unavoidable that an ion exchange process is initiated. As we don’t know any detail of the preparation process, this introduces an uncertainty of the exact aqueous chemistry during the course of a test.
To illustrate this problem, here are the results from calculating the
exchange equilibrium between a sample initially containing 30%
exchangeable charge in form of calcium (70% sodium), and external
NaCl solutions of various concentrations and volumes
In these calculations we assume a sample of density 1.8 g/cm3 with the
same volume as in Mu88 (10.6 cm3), a cation exchange capacity of 0.75
eq/kg, and a Ca/Na selectivity coefficient of 5.
In a main series, we varied the external volume between 50 and 1000 ml
(solid lines). While the solution volume naturally has a significant
influence on the process, it is seen that the initial calcium content
essentially remain for the lowest concentration (0.01 M). In contrast,
for a 1.0 M solution, a significant amount of calcium is exchanged for
all the solution volumes.
The figure also shows a case for sample density 1.2 g/cm3 (dashed line), and a scenario where equilibrium has been obtained twice, with a replacement of the first solution (to a once again pure NaCl solution) (dot-dashed line).
The main lesson from these simulations is that the actual amount of di-valent ions present during a diffusion test depends on many details: the way samples were saturated, volume of external solutions, if and how often solutions were replaced, time, etc. It is therefore impossible to state the exact ion population in any of the tests in Mu88. But, guided by the simulations, it seems very probable that the tests performed at 0.01 M contain a substantial amount of di-valent ions, while those performed at 1.0 M probably resemble more pure sodium systems.
The only information on external solutions in Mu88 is that the
“solution on the low concentration side was changed regularly”
during the course of a test. This implies that the amount of di-valent
cations may not even be constant during the tests.
Uncertainty of diffusion parameters
The diffusion parameters explicitly listed in Mu88 are \(D_e\) and “\(D_a\)”, while it is implicitly understood that they have been obtained by fitting the effective porosity model to outflux data and the measured clay concentration profile in the final state. “\(D_a\)” is thus really the pore diffusivity \(D_p\),7 and relates to \(D_e\) as \(D_e = \epsilon_\mathrm{eff} D_p\), where \(\epsilon_\mathrm{eff}\) is the so-called “effective porosity”. In a previous blog post, we discussed in detail how anion equilibrium concentrations can be extracted from through-diffusion tests, and the results derived there is used extensively in this section.
Rather than fitting the model to the full set of data (i.e. outflux
evolution and final state concentration profile), diffusion parameters
in Mu88 have been extracted in various limits.
Evaluation of \(D_e\) in Mu88
The effective diffusivity was obtained by estimating the steady-state flux, dividing by external concentration difference of the tracer, and multiplying by sample length \begin{equation} D_e = \frac{j^\mathrm{ss}\cdot L}{c^\mathrm{source}}\tag{1} \end{equation}
Here it is assumed that the target reservoir tracer concentration can
be neglected (we assume this
throughout). Eq. 1 is basically eq. 1 in
Mu88
(and
eq. 8 in the earlier blog post), from which we can evaluate the
values of the steady-state flux that was used for the reported values
of \(D_e\) (\(A \approx 7.1\) cm2 denotes sample cross sectional area)
Test
\(D_e\)
\(A\cdot j^\mathrm{ss}/c^\mathrm{source}\)
(\(\mathrm{m^2/s}\))
(ml/day)
1.2/0.01
\(7.7\cdot 10^{-12}\)
0.031
1.2/0.1
\(2.9\cdot 10^{-11}\)
0.118
1.2/1.0
\(1.2\cdot 10^{-10}\)
0.489
1.8/0.01
\(3.3\cdot 10^{-13}\)
0.001
1.8/0.1
\(4.8\cdot 10^{-13}\)
0.002
1.8/1.0
\(4.0\cdot 10^{-12}\)
0.016
The figure below compares the evaluated values of the steady-state
flux with the flux evaluated from the measured target concentration
evolution,8 for samples with nominal dry
density 1.8 g/cm3 (no concentration data was reported for the 1.2
g/cm3 samples)
These plots clearly show that the transition to steady-state is only
resolved properly for the test with highest background concentration
(1.0 M). It follows that the uncertainty of the evaluated steady-state
— and, consequently, of the evaluated \(D_e\) values — increases
dramatically with decreasing background concentration for these
samples.
Evaluation of \(D_p\) in Mu88
Pore diffusivities were obtained in two different ways. One method was to relate the steady-state flux to the clay concentration profile at the end of the test, giving \begin{equation} D_{p,c} = \frac{j^\mathrm{ss}\cdot L}{\phi\cdot\bar{c}(0)} \tag{2} \end{equation}
where \(\bar{c}(0)\) denotes the chloride clay concentration at the interface to the source reservoir. The quantity in eq. 2 is called “\(D_{ac}\)”7 in Mu88, and this equation is essentially the same as eq. 2 in Mu889 (and eq. 10 in the previous blog post). Using the steady-state fluxes, we can back-calculate the values of \(\bar{c}(0)\) used for this evaluation of \(D_{p,c}\)
Test
\(D_{p,c}\)
\(A\cdot j^\mathrm{ss}/c^\mathrm{source}\)
\(\phi\)
\(\bar{c}(0)/c^\mathrm{source}\)
(\(\mathrm{m^2/s}\))
(ml/day)
(-)
(-)
1.2/0.01
\(7.0\cdot 10^{-11}\)
0.031
0.54
0.204
1.2/0.1
\(2.8\cdot 10^{-10}\)
0.118
0.52
0.199
1.2/1.0
\(5.1\cdot 10^{-10}\)
0.489
0.49
0.480
1.8/0.01
\(2.0\cdot 10^{-11}\)
0.001
0.37
0.045
1.8/0.1
\(3.1\cdot 10^{-11}\)
0.002
0.31
0.050
1.8/1.0
\(5.2\cdot 10^{-11}\)
0.016
0.34
0.226
Note that, although we did some calculations to obtain them, the values for \(\bar{c}(0)/c^\mathrm{source}\) in this table are closer to the actual measured raw data (concentrations). We made the calculation above to “de-derive” these values from the reported diffusion coefficients (combining eqs. 1 and 2 shows that \(\bar{c}(0)\) is obtained from the reported parameters as \(\bar{c}(0)/c^\mathrm{source} = D_e/(\phi D_{p,c})\)).
Here are compared the measured concentration profiles for the samples
of nominal density 1.8 g/cm3 and the corresponding slopes used to
evaluate \(D_{p,c}\) (profiles for the 1.2 g/cm3 samples are not
provided in Mu88)
For background concentrations 1.0 M and 0.1 M, the evaluated slope
corresponds quite well to the raw data. For the 0.01 M sample,
however, the match not very satisfactory. I suspect that a detection
limit may have been reached for the analysis of the profile of this
sample. Needless to say, the evaluated value of \(\bar{c}(0)\) is very
uncertain for the 0.01 M sample.
It may also be noted that all measured concentration profiles deviates from linearity near the interface to the source reservoir. This is a general behavior in through-diffusion tests, which I am quite convinced of is related to sample swelling during dismantling, but there are also other suggestedexplanations. Here we neglect this effect and relate diffusion quantities to the linear parts of profiles, but this issue should certainly be treated in a separate discussion.
\(D_p\) was also evaluated in a different way in Mu88, by measuring what we here will call the breakthrough time, \(t_\mathrm{bt}\) (Mu88 call it “time-lag”). This quantity is fairly abstract, and relates to the asymptotic behavior of the analytical expression for the outflux that apply for constant boundary concentrations (we here assume them to be \(c^\mathrm{source}\) and 0, respectively). This expression is displayed in eq. 7 in the previous blog post.
Multiplying the outflux by the sample cross sectional area \(A\) and integrating, gives the accumulated amount of diffused tracers. In the limit of long times, this quantity is, not surprisingly, linear in \(t\) \begin{equation} A\cdot j^\mathrm{ss} \cdot \left(t – \frac{L^2}{6\cdot D_p} \right ) \end{equation}
\(t_\mathrm{bt}\) is defined as the time for which this asymptotic
expression is zero. Determining \(t_\mathrm{bt}\) from the measured
outflux evolution consequently allows for an estimation of \(D_p\) as
\begin{equation}
D_{p,t} = \frac{L^2}{6t_\mathrm{bt}} \tag{3}
\end{equation}
This quantity is called “\(D_{at}\)” in Mu887
(eq. 3 is eq. 3 in Mu88). With another back
calculation we can extract the values of \(t_\mathrm{bt}\) determined
from the raw data
Test
\(D_{p,t}\)
\(t_\mathrm{bt}\)
(\(\mathrm{m^2/s}\))
(days)
1.2/0.01
\(1.4\cdot 10^{-10}\)
3.1
1.2/0.1
\(2.0\cdot 10^{-10}\)
2.2
1.2/1.0
\(3.2\cdot 10^{-10}\)
1.4
1.8/0.01
\(5.0\cdot 10^{-11}\)
8.7
1.8/0.1
\(5.4\cdot 10^{-11}\)
8.0
1.8/1.0
\(7.7\cdot 10^{-11}\)
5.6
These evaluated breakthrough times are indicated
in the flux plots above for samples of
nominal dry density 1.8 g/cm3. For the 0.1 M and 0.01 M
samples it is obvious that this value is very uncertain — without a
certain steady-state flux it is impossible to achieve a certain
breakthrough time. The breakthrough time for the 1.8/1.0 test, on
the other hand, simply appears to be incorrectly evaluated: in terms
of outflux vs. time, the breakthrough time should be the time where
the flux has reached 62% of the steady-state
value.10
As no raw data is reported for the 1.2 g/cm3 tests, the quality of the
evaluated breakthrough times cannot be checked for them. It may be
noted, however, that the evaluated breakthrough times are
significantly shorter in this case as compared with the 1.8 g/cm3
tests. Consequently, while the sampling frequency is high enough to
properly resolve the transient stage of the outflux evolution for the
1.8g/cm3 tests, it must be substantially higher in order to resolve
this stage in the 1.2g/cm3 tests (I guess a rule of thumb is that
sampling frequency must be at least higher than \(1/t_{bt}\)).
In a well conducted study these estimates should be similar; \(D_{p,c}\) and \(D_{p,t}\) are, after all, estimations of the same quantity: the pore diffusivity \(D_p\).7 But here we note a discrepancy of approximately a factor 2 between several values of \(\bar{c}(0)\).
It is difficult to judge generally which of the estimations are more
accurate, but we have seen that for the 1.8/0.1 and 1.8/0.01 tests,
the flux data is not very well resolved, giving a
corresponding uncertainty on the equilibrium concentration estimated
from the breakthrough time. On the other hand, also
the concentration profile is poorly
resolved in the case of 0.01 M at 1.8 g/cm3.
However, in cases where the value of \(\bar{c}(0)/c^\mathrm{source}\) is substantial (as for the 1.8/1.0 test and, reasonably, for all tests at 1.2 g/cm3), we expect the estimation directly from the concentration profile to be accurate and robust (as for the 1.8 g/cm3 test at high NaCl concentration). For the 1.2 g/cm3 samples we cannot say much more than this, since Mu88 don’t provide the concentration raw data. For the 1.8/1.0 test, however, we can continue the analysis by fitting the model to all available data.
Re-evaluation by fitting to the full data set
Note that all evaluations in Mu88 are based on making an initial estimation of the steady-state flux, giving \(D_e\) (eq. 1). This value of \(D_e\) (or \(j^{ss}\)) is thereafter fixed in the subsequent estimation of \(D_{p,c}\) (eq. 2). Likewise, an estimation of the steady-state flux is required for estimating the breakthrough time. Here is an animation showing the variation of the model when transitioning from the value of the pore diffusivity estimated from breakthrough time (\(7.7\cdot 10^{-11}\) m2/s), to the value estimated from concentration profile (\(5.2\cdot 10^{-11}\) m2/s) for the 1.8/1.0 test, keeping the steady-state flux fixed at the initial estimation
Note that the axes for the flux is on top (time) and to the right (accumulation rate). This animation confirms that the diffusivity evaluated from breakthrough time in Mu88 gives a way too fast process: the slope of the steady-state concentration profile is too small, and the outflux evolution has a too short transient stage. On the other hand, using the diffusivity estimated from the concentration profiles still doesn’t give a flux that fit very well. The problem is that this fitting is performed with a fixed value of the steady-state flux. By instead keeping the slope fixed at the experimental values, while varying diffusivity (and thus steady state flux), we get the following variation
This animation shows that the model can be fitted well to all data (at least for the 1.8/1.0 test). The problem with the evaluation in Mu88 is that it assumes the steady-state to be fully reached at the later stages of the test. As the above fitting procedure shows, this is only barely true. The experiments could thus have been designed better by conducting them longer, in order to better sample the steady-state phase (and the steady-state flux should have been fitted to the entire data set). Nevertheless, for this sample, the steady-state flux obtained by allowing for this parameter to vary is only slightly different from that used in Mu88 (17.5 rather than 16.3 \(\mathrm{\mu}\)l/day, corresponding to a change of \(D_p\) from \(5.2\cdot10^{-11}\) to \(5.6\cdot10^{-11}\) m2/s). Moreover, this consideration should not be a problem for the 1.2 g/cm3 tests, if they were conducted for as long time as the 1.8 g/cm3 tests, because steady-state is reached much faster (in those tests, sampling frequency may instead be a problem, as discussed above).
As we were able to fit the full model to all data, we conclude that the value of \(\bar{c}(0)/c^\mathrm{source}\) obtained from \(D_{p,c}\) is probably the more robust estimation11, and that there appears to be a problem with how the breakthrough times have been determined. For the 1.8 g/cm3 samples we have demonstrated that this is the case, for the 1.2 g/cm3 we can only make an educated guess that this is the case.
Summary and verdict
We have seen that the results on chloride diffusion in Mu88 suffer from uncertainty from several sources:
The “MX-80” material is not that well defined
Densities vary substantially for samples at the same nominal density
Without knowledge of e.g water saturation procedures and solution volumes, it is impossible to estimate the proper ion population during the course of a test
It is, however, highly likely that tests performed at low NaCl concentrations contain substantial amounts of di-valent ions, while those at high NaCl concentration are closer to being pure sodium systems.
The reported diffusivities give a corresponding uncertainty in the chloride equilibrium concentrations of about a factor of 2. While some tests essentially have a too high noise level to give certain estimations, the problem for the others seems to stem from the estimation of breakthrough times.
Here is an attempt to encapsulate the above information in an
updated plot for the chloride equilibrium data in Mu88
The colored squares represent “confidence areas” based on the variation within each nominal density (horizontally), and on the variation of \(\bar{c}(0)/c^\mathrm{source}\) from the two reported values on pore diffusivity7 (vertically). The limits of these rectangles are simply the 95% confidence interval, based on these variations, and assuming a normal distribution.
Data points put within parentheses are estimations judged to be
improper (based on either re-evaluation of the raw data, or informed
guesses).
From the present analysis my decision is to not use the data
from Mu88 to e.g. validate models for anion exclusion. Although there
seems to be nothing fundamentally wrong with how these test were
conducted, they suffer from so many uncertainties of various sources
that I judge the data to not contribute to quantitative process
understanding.
[2] MX-80 is not only a brand name, but also
a band
name.
[3] This report is “Bentonite Mineralogy” by L. Carlson (Posiva WR 2004-02), but it appears to not be included in the INIS database. It can, however, be found with some elementary web searching.
[4] It’s interesting to note that the cation exchange capacity of
“MX-80” remains more or less constant, while the montmorillonite
content has some variation. This implies that the montmorillonite
layer charge varies (and is negatively correlated with montmorillonite
content). Could it be that the manufacturer has a specified cation
exchange capacity as requirement for this product?
[5] To convert porosity to
dry density, I used \(\rho_d = \rho_s\cdot(1-\phi)\), with solid grain
density \(\rho_s = 2.75\) g/cm3.
[6] A speculation is that the uncertainty stems from the measurement procedure, as this was done on smaller sections of the full samples. It is not specified in Mu88 what the reported porosity represent, but it is reasonable to assume that it is the average of all sections of a sample.
[8] These values were not tabulated, but I have read
them off from the graphs in Mu88.
[9] Mu88 use the
concentration based on the total volume in their expression, while
\(\bar{c}\) is
defined in terms of water volume (water mass,
strictly). Eq.2 therefore contains the physical
porosity. In their concentration profile plots, however, Mu88 use
\(\bar{c}\) as variable (called \(c_{pw}\) — the “concentration in the pore
water”)
[10] Plugging the breakthrough time \(L^2/6D_p\) into the expression for the flux gives
I find it amusing that this value is close to the reciprocal golden ratio (0.618033…). Finding the breakthrough time from a flux vs. time plot thus corresponds (approximately) to splitting the y-axis according to the golden ratio.
[11] Note that the actual evaluated values of $D_{p,c}$ in Mu88 still may be uncertain, because they also depend on the values of the steady-state flux, which we have seen were not optimally evaluated.
Researchers traditionally measure sorption on montmorillonite in batch tests, where a small amount of solids is mixed with a tracer-spiked solution (typical solid-to-liquid ratios are \(\sim 1 – 10\) g/l). After equilibration, solids and solution are usually separated by centrifugation and the supernatant is analyzed.
This procedure evidently counts tracer cations that reside in diffuse layers as sorbed. But tracer ions may also sorb due to other mechanisms, in particular due to bonding on specific surface hydroxyl groups, on the edges of individual montmorillonite layers. These different types of “sorption” are in the clay literature usually referred to as “cation exchange” and “surface complexation”, respectively.
The amount of tracer “sorbed” in the ways just described is
quantified by the distribution coefficient \(K_d\), defined as
\begin{equation}
s = K_d\cdot c_\mathrm{eq}
\end{equation}
where \(s\) denotes the amount of tracers “on the solids”, and
\(c_\mathrm{eq}\) is the corresponding equilibrium concentration in the
aqueous phase. As the amount “on the solids” can be inferred from
the amount of tracers that has been removed from the initial solution,
we can evaluate \(K_d\) from
where \(c_\mathrm{init}\) is the initial tracer concentration
(i.e. before adding the clay), \(c_\mathrm{final}\) is the tracer
concentration in the supernatant, \(V_\mathrm{sol}\) is the solution
volume, and \(m_s\) is the mass of the solids.
If the purpose of a study is solely to quantify the amount of tracer
“on the solids”, it is adequate to define sorption as including
both “cation exchange” and “surface complexation”, and to use
\(K_d\) as the measure of this sorption. However, if our main concern is
to describe transport in compacted bentonite, \(K_d\) is a
rather blunt tool, since it quantifies both ions that dominate the
transport capacity (“cation exchange”), and ions that are immobile,
or at least contribute to an actual delay of diffusive fluxes
(“surface complexation”).
Moreover, when evaluating \(K_d\) in batch tests, or when using this
parameter in models, authors assume that the solids are in equilibrium
locally with a bulk water phase. But there is no compelling evidence
that such a phase exists in compacted water saturated bentonite. On
the contrary,
severalobservationsstrongly suggest
that compacted bentonite lacks significant amounts of bulk
water. This, in turn, suggests that \(K_d\) actually quantifies the
equilibrium between a bentonite sample and an external
solution.
Indeed, even in batch tests is the final concentration measured in a solution (the supernatant) separated from the clay (the sediment), as a consequence of the centrifugation, as illustrated here:
This figure also illuminates additional and perhaps more subtle complications when evaluating \(K_d\) from batch tests. Firstly, such values are implicitly assumed independent of “sample” density. There are, however, arguments for that \(K_d\) in general depends on density, as will be explored below. The question is then to what density range we can apply batch test values when modeling compacted systems, or if they can be applied at all. Note that the “sample” that is measured on in a batch test (see figure) has a more or less well-defined density. But sediment densities are, to my knowledge, never investigated in these types of studies.1
Secondly, it could be questioned if the supernatant have had time to
equilibrate with the sediment, i.e whether
\(c_\mathrm{final} = c_\mathrm{eq}\). Instead, as far as I know,
researchers routinely assume that the equilibrium established prior to
centrifugation remains.
In the following, we use
the homogeneous mixture model
to analyze in more detail the nature of \(K_d\) in compacted bentonite.
Kd in the homogeneous mixture model
As usual when analyzing bentonite with the homogeneous mixture model, we assume an external solution in contact with a homogeneous bentonite domain at a specific density (water-to-solid mass ratio \(w\)). The bentonite and the external solution are separated via a semi-permeable component, which allows for the passage of water and ions, but does not allow for the passage of clay (symbols are explained below):
This model resembles the alternative test set-up for determining \(K_d\) in compacted systems used by Van Loon and Glaus (2008), where the clay is contained in a sample holder, and the tracer is supplied through a filter from an external circulating solution. This approach has the advantages that the state of the clay is controlled throughout the test (which, e.g., allows for investigating how \(K_d\) depends on density), and that the equilibration process is better controlled (avoiding the possible disruptive procedure of centrifugation). The obvious disadvantage is that equilibration — being diffusion controlled — may take a long time.
When applying the homogeneous mixture model in
earlierblog posts,
we have assumed “simple” ions, which contribute to the ion
population of the clay only in terms of the interlayer concentration,
\(c^\mathrm{int}\). This concentration quantifies the amount of mobile
ions involved in establishing Donnan equilibrium between clay and
external solutions. But many “non-simple” ions actually do seem to
be immobilized/delayed by also associate with surfaces
(\(\mathrm{H}^+\), \(\mathrm{Ni}^{2+}\), \(\mathrm{Zn}^{2+}\),
\(\mathrm{Co}^{2+}\), \(\mathrm{P_2O_7^{4-}}, …\)). For a more general
description, we therefore extend the homogeneous mixture model with a
second contribution to the ion population: \(s^\mathrm{int}\) (ions per
unit mass).
Using the traditional terminology, the ions quantified by
\(c^\mathrm{int}\) are to be identified as “sorbed by ion exchange”,
and those quantified by \(s^\mathrm{int}\) as “sorbed by surface
complexation”. But since the ion exchange process does not immobilize
ions and primarily should be
associated with Donnan equilibrium,
we want to avoid referring to them as “sorbed”. Also, with the
traditional terminology, all ions in the homogeneous mixture
model are described as “sorbed”, which obviously not is very useful.
We therefore introduce different terms, and refer to the ions
quantified by \(c^\mathrm{int}\) as aqueous interlayer species,
and to the ions quantified by \(s^\mathrm{int}\) as truly sorbed
ions. With this terminology, the term “sorption” puts emphasis on
ions being immobile.2 Moreover, the description now
also applies to anions, without having to refer to them as
e.g. “sorbed by ion exchange”.
In analogy with the traditional diffusion-sorption model, we assume a
linear relation between \(s^\mathrm{int}\) and
\(c^\mathrm{int}\)
where \(\Lambda\) is a distribution coefficient quantifying the relation
between the amount of aqueous species in the interlayer domain
and amount of truly sorbed substance.3
The amount of an aqueous species in the homogeneous mixture model is \(V_p\cdot c^\mathrm{int}\), where \(V_p\) is the total pore volume. The total amount of an ion per unit mass is thus \(V_p\cdot c^\mathrm{int}/m_s + s^\mathrm{int}\), where \(m_s\), as before, denotes total solid mass.
To get an expression for \(K_d\) in the homogeneous mixture model, we
must associate ions “on the solids” (\(s\)) with the concentration in
the external solution. Here we choose the simplest way to do this, and
write
which implies that we define all ions in the bentonite sample to be “on the solids”. To be fully consistent, we should perhaps subtract the contribution expected to be found in the clay if it behaved like a conventional porous system (\(V_p\cdot c^\mathrm{ext}/m_s\)). But, since we are mostly interested in the limit of small \(V_p/m_s\), this contribution can be thought of as becoming arbitrary small, and we therefore don’t bother with including it in the formulas. In any case, this “conventional porewater” contribution would simply give an extra term \(-w/\rho_w\) in the equations we are about to derive, and can be included if desired.
Using eqs. 1
and 2, we get the expression for \(K_d\) in
the homogeneous mixture model
where we also have used the definition of the ion equilibrium
coefficient \(\Xi = c^\mathrm{int}/c^\mathrm{ext}\), and utilized that
\(V_p/m_s = w/\rho_w\), where \(\rho_w\) is the density of
water.4
A full analysis of eq. 3 is a major task, but
a few things are immediately clear:
\(K_d\) generally has two contributions: one from Donnan
equilibrium (\(w\cdot\Xi/\rho_w\)) and one from true
sorption (\(\Lambda\cdot \Xi\)). Using the traditional terminology,
these contributions correspond for cations to “sorption by ion
exchange” and “sorption by surface complexation”, respectively. But
note that eq. 3 is valid also for anions.
For a simple cation (\(\Lambda = 0\)), \(K_d\) merely quantifies the
aqueous interlayer concentration.5As we have discussed earlier,
\(K_d\) quantifies in this case a type of enhancement of the transport
capacity. I think it is unfortunate that a mechanism that dominates
the mass transfer capacity traditionally is labeled “sorption”.
For cations with \(\Lambda \neq 0\), \(K_d\) is not a measure of
true sorption, because we always expect a significant Donnan
contribution. In this case \(K_d\) quantifies a mixture of transport
enhancing and transport inhibiting mechanisms. Clearly, it is
unsatisfactory to use the term “sorption” for mechanisms that both
enhance and reduce transport capacity (at least when the objective
is a transport description).
For simple anions, the above expression gives a positive value
for \(K_d\). Traditionally, the \(K_d\) concept has not been applied to
these types of ions, and e.g. chloride is often
described as “non-sorbing”,
with \(K_d =0\). Since \(\Xi \rightarrow 0\) as \(w \rightarrow 0\)
generally for anions, this result (\(K_d = 0\)) is recovered in this
limit.6
Kd for simple cations
We end this post by examining expressions for \(K_d\) for simple cations in some specific cases. In the following we consequently assume \(\Lambda = 0\), and this section relies heavily on the ion equilibrium framework in the homogeneous mixture model, with the main relation
where \(z\) is the charge number of the ion,
\(\Gamma \equiv \gamma^\mathrm{ext}/\gamma^\mathrm{int}\) is an activity
coefficient ratio, and \(f_D = e^\frac{F\psi^\star}{RT}\) is the
so-called Donnan factor, with \(\psi^\star\) (\(<0\)) being the Donnan
potential.
Simple cation tracers in a 1:1 system
We assume a bentonite sample at water-to-solid mass ratio \(w\) in
equilibrium with an external 1:1 solution (e.g. NaCl) of concentration
\(c^\mathrm{bgr}\). The Donnan factor is in this case, in the limit
\(c^\mathrm{bgr} \ll c_\mathrm{IL}\)7
where \(CEC\) is the cation exchange capacity, and \(F\) is the Faraday constant (1 eq/mol). We furthermore assume the presence of a mono-valent cation tracer, which, by definition, does not influence \(f_D\). The ion equilibrium coefficient for this tracer is (from eq. 4)
Note that \(K_d\) for a mono-valent ion in a 1:1 system does not explicitly depend on density (eq. 5), while \(K_d\) for a di-valent ion diverges as \(w\rightarrow 0\) (eq. 6). In contrast, \(K_d\) in a 2:1 system has no explicit density dependence for di-valent tracers (eq. 8), while \(K_d\) vanishes for a mono-valent tracer in the limit \(w \rightarrow 0\) (eq. 7).
These results imply that we expect \(K_d\) to generally depend on sample density in systems where the charge number of the tracer ions differs from that of the cation of the background electrolyte. It may therefore not be appropriate to use values of \(K_d\) evaluated in batch-type tests for analyzing compacted systems.
Note also that \(K_d\) may have significant density dependence also in cases where the present analysis gives no explicit \(w\)-dependence on \(K_d\). This was demonstrated e.g. by Van Loon and Glaus (2008) for cesium tracers in sodium dominated bentonite. Interpreted in terms of the homogeneous mixture model, their results show that the interlayer activity coefficients vary significantly with density. In particular, the results imply either that the interlayer activity coefficient for cesium becomes small (\(\Gamma_\mathrm{Cs} \gg 1\)), or that the interlayer activity coefficient for sodium becomes large (\(\Gamma_\mathrm{Na} \ll 1\)), in the high density limit.
[1] A sediment density is, reasonably, related to e.g. initial solid-to-water ratio and to the details of the centrifugation procedure.
[2] I am not very happy with this
terminology, but we need a way to distinguish this type of sorption
from how the term “sorption” is used in the bentonite literature,
where it nowadays essentially refers to
the process of taking up an ion from a bulk water phase to some other phase.
This is the reason for why there are so many quotation marks around
the word “sorption” in the text.
[3] I don’t know if
this is a valid assumption, but it seems like the natural starting
point.
[4] The presence of water density in the formulas reflects
the fact that we are using molar units (substance per unit
volume), which is natural, as \(K_d\) typically has units of
volume per mass. How to associate a density to water in the
homogeneous mixture model is a bit subtle, and we don’t focus on
that aspect here (it may be the issue of future posts). In the
presented formulas \(\rho_w\) can rather be viewed as a unit
conversion factor.
[5] When
\(\Lambda = 0\), we can rearrange eq. 3
as
where \(\rho_d\) is dry density, \(\phi\) is porosity, and \(\kappa\) was defined as a scaled, dimensionless version of \(K_d\) by Gimmi and Kosakowski (2011), discussed in a previous blog post. Interpreted using the homogeneous mixture model, \(\kappa\) is thus simply the ion equilibrium coefficient for simple cations.
[6] By including the “conventional porewater”
contribution in the definition of \(K_d\), as discussed earlier, we
get for these types of anions
This is typically a negative quantity, and quantifies anion exclusion, in the Schofield sense of the term. We have, also with this definition, that \(K^\prime_d \rightarrow 0\) as \(w \rightarrow 0\).
[7] We assume
\(c^\mathrm{bgr} \ll c_\mathrm{IL}\) in this and all following
cases. For compacted bentonite \(c_\mathrm{IL}\) is of the order of
several molar, and the derived approximations are thus valid for
“typical” background concentrations (\(< 1\) M). Also, for an
arbitrary value of \(c^\mathrm{bgr}\), one can in principle always
choose a sufficiently low value of \(w\) to satisfy
\(c^\mathrm{bgr} \ll c_\mathrm{IL}\).
[8] If the selectivity coefficient is identified with
that derived in
Birgersson
(2017).
Recently, we discussed reported equilibrium chloride concentrations in sodium dominated bentonite, and identified a need to assess the individual studies. As most data is obtained from through-diffusion experiments, we here take a general look at how anion equilibrium is a part of the through-diffusion set-up, and how we can use reported model parameters to extract the experimentally accessible equilibrium concentrations.
We define the experimentally accessible concentration of a chemical
species in a bentonite sample as
where \(n\) is the total amount of the
species,1 and \(m_{w}\) is
the total water mass in the
clay.2 It should be clear
that \(\bar{c}\), which we will refer to as the clay concentration, is
accessible without relying on any particular model concept.
An equilibrium concentration is defined as the corresponding clay concentration (i.e. \(\bar{c}\)) of a species when the clay is in equilibrium with an external solution with species concentration \(c^\mathrm{ext}\). A convenient way to express this equilibrium is in terms of the ratio \(\bar{c}/c^\mathrm{ext}\).
The through-diffusion set-up
A through-diffusion set-up consists of a (bentonite) sample sandwiched between a source and a target reservoir, as illustrated schematically here (for some arbitrary time):
The sample length is labeled \(L\), and we assume the sample to be
initially empty of the diffusing species. A test is started by adding
a suitable amount of the diffusing species to the source
reservoir. Diffusion through the bentonite is thereafter monitored by
recording the concentration evolution in the target
reservoir,3 giving
an estimation of the flux out of the sample (\(j^\mathrm{out}\)). The
clay concentration for anions is typically lower than the
corresponding concentration in the source reservoir.
Although a through-diffusion test is not in full equilibrium (by definition), local equilibrium prevails between clay and external solution4 at the interface to the source reservoir (\(x=0\)). Thus, even if the source concentration varies, we expect the ratio \(\bar{c}(0)/c^\mathrm{source}\) to stay constant during the course of the test.5
The effective porosity diffusion model
Our primary goal is to extract the concentration ratio \(\bar{c}(0)/c^\mathrm{source}\) from reported through-diffusion parameters. These parameters are in many anion studies specific to the “effective porosity” model, rather than being accessible directly from the experiments. We therefore need to examine this particular model.
The effective porosity model divides the pore space into a bulk water domain and a domain that is assumed inaccessible to anions. The porosity of the bulk water domain is often referred to as the “effective” or the “anion-accessible” porosity, and here we label it \(\epsilon_\mathrm{eff}\).
Anions are assumed to diffuse in the bulk water domain according to
Fick’s first law
where \(D_p\) is the pore diffusivity in the bulk water phase. This
relation is alternatively expressed as
\(j = -D_e \cdot \nabla c^\mathrm{bulk}\), which defines the effective
diffusivity \(D_e = \epsilon_\mathrm{eff} \cdot D_p\).
Diffusion is assumed to be the only mechanism altering the
concentration, leading to Fick’s second law
where \(\phi\) is the physical porosity of the sample. Since a bulk water concentration varies continuously across interfaces to external solutions, we have \(c^\mathrm{bulk}(0) = c^\mathrm{source}\) at the source reservoir, giving
This equation shows that the effective porosity parameter quantifies
the anion equilibrium concentration that we want to extract. That is
not to say that the model is valid (more on that later), but that we
can use eq. 4 to translate reported model parameters to an
experimentally accessible quantity.
In principle, we could finish the analysis here, and use eq. eq. 4 as our main result. But most researchers do not evaluate the effective porosity in the direct way suggested by this equation (they may not even measure \(\bar{c}\)). Instead, they evaluate \(\epsilon_\mathrm{eff}\) from a fitting procedure that also includes the diffusivity as a parameter. It is therefore fruitful to also include the transport aspects of the through-diffusion test in our analysis.
From closed-cell diffusion tests, we know that the clay concentration evolves according to Fick’s second law, both for many cations and anions. We will therefore take as an experimental fact that \(\bar{c}\) evolves according to
This equation defines the diffusion coefficient \(D_\mathrm{macr.}\), which should be understood as an empirical quantity.
Combining eqs. 3 and 2 shows that \(D_p\) governs the evolution of \(\bar{c}\) in the effective porosity model (if \(\epsilon_\mathrm{eff}/\phi\) can be considered a constant). A successful fit of the effective porosity model to experimental data thus provides an estimate of \(D_\mathrm{macr.}\) (cf. eq. 5), and we may write
With the additional assumption of constant reservoir concentrations,
eq. 2 has a relatively simple analytical solution, and the
corresponding outflux reads
where \(j^\mathrm{ss}\) is the steady-state flux. In steady-state,
\(c^\mathrm{bulk}\) is distributed linearly across the sample, and we
can express the gradient in eq. 1 using the reservoir
concentrations, giving
Treating \(j^\mathrm{ss}\) as an empirical parameter (it is certainly
accessible experimentally), and using eq. 6, we get another expression
for \(\epsilon_\mathrm{eff}\) in terms of experimentally accessible
quantities
This relation (together with eqs. 4 and 6) demonstrates that if we fit eq. 7 using \(D_p\) and \(j^\mathrm{ss}\) as fitting parameters, the equilibrium relation we seek is given by
This procedure may look almost magical, since any explicit reference to the effective porosity model has now disappeared; eq. 10 can be viewed as a relation involving only experimentally accessible quantities.
But the validity of eq. 10 reflects the empirical fact that the (steady-state) flux can be expressed using the gradient in \(\bar{c}\) and the physical porosity. The effective porosity model can be successfully fitted to anion through-diffusion data simply because it complies with this fact. Consequently, a successful fit does not validate the effective porosity concept, and essentially any description for which the flux can be expressed as \(j = -\phi\cdot D_p \cdot \nabla\bar{c}\) will be able to fit to the data.
We may thus consider a generic model for which eq. 5 is valid and for which a steady-state flux is related to the external concentration difference as
where \(\beta\) is an arbitrary constant. Fitting such a model, using \(\beta\) and \(D_p\) as parameters, will give an estimate of \(\bar{c}(0)/c^\mathrm{source}\) (\(=\beta / \phi\)).
Note that the system does not have to reach steady-state — eq. 11 only states how the model relates a steady-state flux to the reservoir concentrations. Moreover, the model being fitted is generally numerical (analytical solutions are rare), and may account for e.g. possible variation of concentrations in the reservoirs, or transport in the filters connecting the clay and the external solutions.
The effective porosity model emerges from this general description by interpreting \(\beta\) as quantifying the volume of a bulk water phase within the bentonite sample. But \(\beta\) can just as well be interpreted e.g. as an ion equilibrium coefficient (\(\phi\cdot \Xi = \beta\)), showing that this description also complies with the homogeneous mixture model.
Additional comments on the effective porosity model
The effective porosity model can usually be successfully fitted to anion through-diffusion data (that’s why it exists). The reason is not because the data behaves in a manner that is difficult to capture without assuming that anions are exclusively located in a bulk water domain, but simply because this model complies with eqs. 5 and 11. We have seen that also the homogeneous mixture model — which makes the very different choice of having no bulk water at all within the bentonite — will fit the data equally well: the two fitting exercises are equivalent, connected via the parameter identification \(\epsilon_\mathrm{eff} \leftrightarrow \phi\cdot\Xi\).
Perhaps even more remarkable is that authors frequently treat the effective porosity model as were it some version of the traditional diffusion-sorption model. This is often done by introducing a so-called rock capacity factor \(\alpha\) — which can take on the values \(\alpha = \phi + \rho\cdot K_d\) for cations, and \(\alpha = \epsilon_\mathrm{eff}\) for anions — and write \(D_e = \alpha D_a\), where \(D_a\) is the “apparent” diffusion coefficient. The reasoning seems to go something like this: since the parameter in the governing equation in one model can be written as \(D_e/\epsilon_\mathrm{eff}\), and as \(D_e/(\phi + \rho\cdot K_d)\) in the other, one can view \(\epsilon_\mathrm{eff}\) as being due to negative sorption (\(K_d < 0\)).
But such a mixing of completely different mechanisms (volume restriction vs. sorption) is just a parameter hack that throws most process understanding out the window! In particular, it hides the fact that the effective porosity and diffusion-sorption models are incompatible: their respective bulk water domains have different volumes. Furthermore, this lumping together of models has led to that anion diffusion coefficients routinely are reported as “apparent”, although they are not; the underlying model contains a pore diffusivity (eq. 2). As I have stated before, the term “apparent” is supposed to convey the meaning that what appears as pure diffusion is actually the combined result of diffusion, sorption, and immobilization. Sadly, in the bentonite literature, “apparent diffusivity” often means “actual diffusivity”.
Footnotes
[1] For anions, the total amount is relatively easy to measure by e.g. aqueous extraction. Cations, on the other hand, will stick to the clay, and need to be exchanged with some other type of cation (not initially present). In any case, the total amount of a species (\(n\)) can in principle be obtained experimentally, in an unambiguous manner.
[2] Another reasonable choice would be to divide by the total sample volume.
[3] If the test is designed as to have a significant change of the source concentration, it is a good idea to also measure the concentration evolution in this reservoir.
[5] Provided that the rest of the aqueous chemistry remains constant, which is not always the case. For instance, cation exchange may occur during the course of the test, if the set-up involves more than one type of cation, and there may be ongoing mineral dissolution.