Measuring Kd
Researchers traditionally measure sorption on montmorillonite in batch tests, where a small amount of solids is mixed with a tracer-spiked solution (typical solid-to-liquid ratios are \(\sim 1 – 10\) g/l). After equilibration, solids and solution are usually separated by centrifugation and the supernatant is analyzed.
This procedure evidently counts tracer cations that reside in diffuse layers as sorbed. But tracer ions may also sorb due to other mechanisms, in particular due to bonding on specific surface hydroxyl groups, on the edges of individual montmorillonite layers. These different types of “sorption” are in the clay literature usually referred to as “cation exchange” and “surface complexation”, respectively.
The amount of tracer “sorbed” in the ways just described is quantified by the distribution coefficient \(K_d\), defined as
\begin{equation} s = K_d\cdot c_\mathrm{eq} \end{equation}
where \(s\) denotes the amount of tracers “on the solids”, and \(c_\mathrm{eq}\) is the corresponding equilibrium concentration in the aqueous phase. As the amount “on the solids” can be inferred from the amount of tracers that has been removed from the initial solution, we can evaluate \(K_d\) from
\begin{equation} K_d = \frac{\left ( c_\mathrm{init} – c_\mathrm{final} \right ) \cdot V_\mathrm{sol}} {c_\mathrm{final}\cdot m_\mathrm{s}} \end{equation}
where \(c_\mathrm{init}\) is the initial tracer concentration (i.e. before adding the clay), \(c_\mathrm{final}\) is the tracer concentration in the supernatant, \(V_\mathrm{sol}\) is the solution volume, and \(m_s\) is the mass of the solids.
If the purpose of a study is solely to quantify the amount of tracer “on the solids”, it is adequate to define sorption as including both “cation exchange” and “surface complexation”, and to use \(K_d\) as the measure of this sorption. However, if our main concern is to describe transport in compacted bentonite, \(K_d\) is a rather blunt tool, since it quantifies both ions that dominate the transport capacity (“cation exchange”), and ions that are immobile, or at least contribute to an actual delay of diffusive fluxes (“surface complexation”).
A good illustration of this problem is the traditional diffusion-sorption model, which incorrectly assumes that all ions quantified by \(K_d\) are immobilized. In earlier blog posts, we have discussed the consequences of this model assumption, and the empirical evidence against it. A complication when discussing sorption is that researchers often “measure” \(K_d\) by fitting the traditional diffusion-sorption model to data — although the model is not valid for compacted bentonite.
Moreover, when evaluating \(K_d\) in batch tests, or when using this parameter in models, authors assume that the solids are in equilibrium locally with a bulk water phase. But there is no compelling evidence that such a phase exists in compacted water saturated bentonite. On the contrary, several observations strongly suggest that compacted bentonite lacks significant amounts of bulk water. This, in turn, suggests that \(K_d\) actually quantifies the equilibrium between a bentonite sample and an external solution.
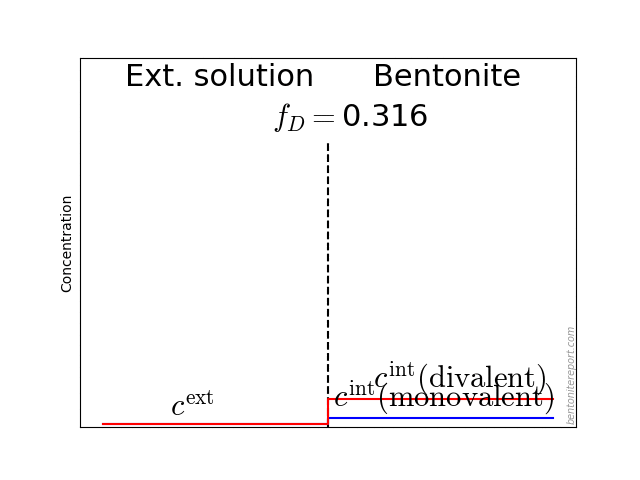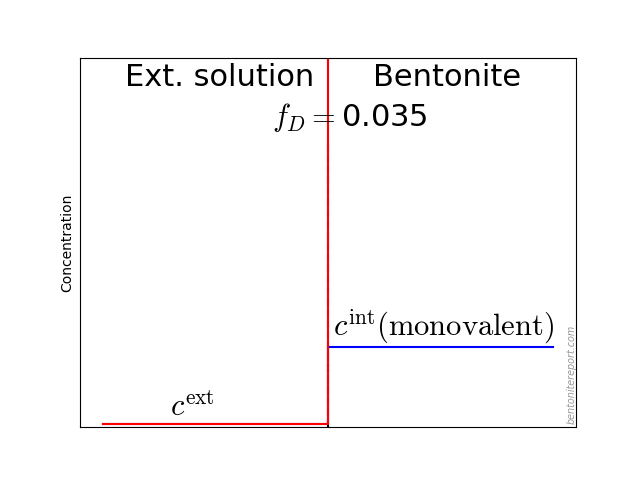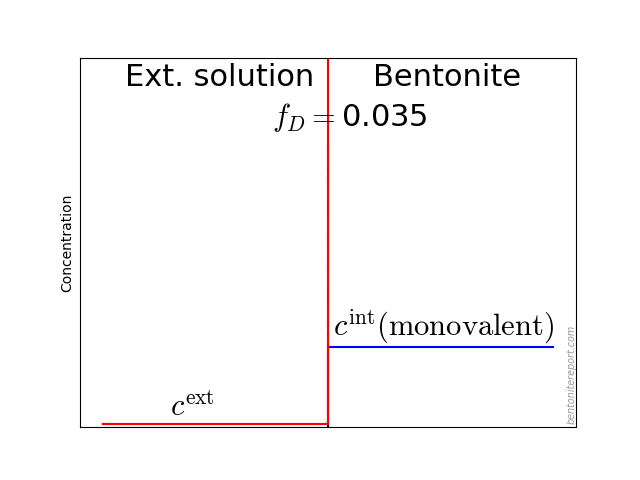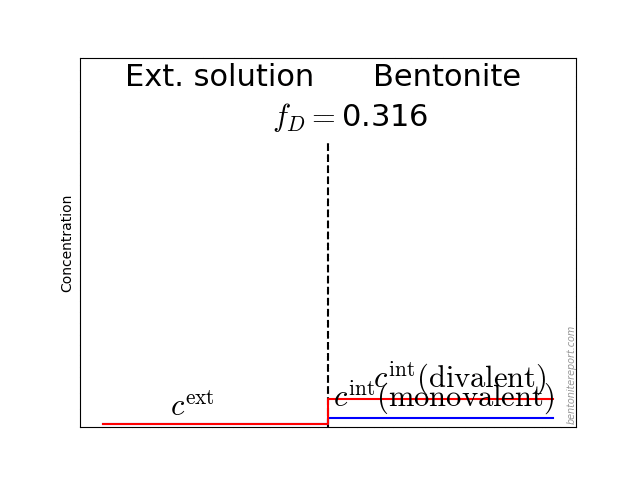
Indeed, even in batch tests is the final concentration measured in a solution (the supernatant) separated from the clay (the sediment), as a consequence of the centrifugation, as illustrated here:

This figure also illuminates additional and perhaps more subtle complications when evaluating \(K_d\) from batch tests. Firstly, such values are implicitly assumed independent of “sample” density. There are, however, arguments for that \(K_d\) in general depends on density, as will be explored below. The question is then to what density range we can apply batch test values when modeling compacted systems, or if they can be applied at all. Note that the “sample” that is measured on in a batch test (see figure) has a more or less well-defined density. But sediment densities are, to my knowledge, never investigated in these types of studies.1
Secondly, it could be questioned if the supernatant have had time to equilibrate with the sediment, i.e whether \(c_\mathrm{final} = c_\mathrm{eq}\). Instead, as far as I know, researchers routinely assume that the equilibrium established prior to centrifugation remains.
In the following, we use the homogeneous mixture model to analyze in more detail the nature of \(K_d\) in compacted bentonite.
Kd in the homogeneous mixture model
As usual when analyzing bentonite with the homogeneous mixture model, we assume an external solution in contact with a homogeneous bentonite domain at a specific density (water-to-solid mass ratio \(w\)). The bentonite and the external solution are separated via a semi-permeable component, which allows for the passage of water and ions, but does not allow for the passage of clay (symbols are explained below):

This model resembles the alternative test set-up for determining \(K_d\) in compacted systems used by Van Loon and Glaus (2008), where the clay is contained in a sample holder, and the tracer is supplied through a filter from an external circulating solution. This approach has the advantages that the state of the clay is controlled throughout the test (which, e.g., allows for investigating how \(K_d\) depends on density), and that the equilibration process is better controlled (avoiding the possible disruptive procedure of centrifugation). The obvious disadvantage is that equilibration — being diffusion controlled — may take a long time.
When applying the homogeneous mixture model in earlier blog posts, we have assumed “simple” ions, which contribute to the ion population of the clay only in terms of the interlayer concentration, \(c^\mathrm{int}\). This concentration quantifies the amount of mobile ions involved in establishing Donnan equilibrium between clay and external solutions. But many “non-simple” ions actually do seem to be immobilized/delayed by also associate with surfaces (\(\mathrm{H}^+\), \(\mathrm{Ni}^{2+}\), \(\mathrm{Zn}^{2+}\), \(\mathrm{Co}^{2+}\), \(\mathrm{P_2O_7^{4-}}, …\)). For a more general description, we therefore extend the homogeneous mixture model with a second contribution to the ion population: \(s^\mathrm{int}\) (ions per unit mass).
Using the traditional terminology, the ions quantified by \(c^\mathrm{int}\) are to be identified as “sorbed by ion exchange”, and those quantified by \(s^\mathrm{int}\) as “sorbed by surface complexation”. But since the ion exchange process does not immobilize ions and primarily should be associated with Donnan equilibrium, we want to avoid referring to them as “sorbed”. Also, with the traditional terminology, all ions in the homogeneous mixture model are described as “sorbed”, which obviously not is very useful.
We therefore introduce different terms, and refer to the ions quantified by \(c^\mathrm{int}\) as aqueous interlayer species, and to the ions quantified by \(s^\mathrm{int}\) as truly sorbed ions. With this terminology, the term “sorption” puts emphasis on ions being immobile.2 Moreover, the description now also applies to anions, without having to refer to them as e.g. “sorbed by ion exchange”.
In analogy with the traditional diffusion-sorption model, we assume a linear relation between \(s^\mathrm{int}\) and \(c^\mathrm{int}\)
\begin{equation} s^\mathrm{int} = \Lambda\cdot c^\mathrm{int} \tag{1} \end{equation}
where \(\Lambda\) is a distribution coefficient quantifying the relation between the amount of aqueous species in the interlayer domain and amount of truly sorbed substance.3
The amount of an aqueous species in the homogeneous mixture model is \(V_p\cdot c^\mathrm{int}\), where \(V_p\) is the total pore volume. The total amount of an ion per unit mass is thus \(V_p\cdot c^\mathrm{int}/m_s + s^\mathrm{int}\), where \(m_s\), as before, denotes total solid mass.
To get an expression for \(K_d\) in the homogeneous mixture model, we must associate ions “on the solids” (\(s\)) with the concentration in the external solution. Here we choose the simplest way to do this, and write
\begin{equation} s = \frac{V_p\cdot c^\mathrm{int}}{m_s} + s^\mathrm{int} = K_d\cdot c^\mathrm{ext} \tag{2} \end{equation}
which implies that we define all ions in the bentonite sample to be “on the solids”. To be fully consistent, we should perhaps subtract the contribution expected to be found in the clay if it behaved like a conventional porous system (\(V_p\cdot c^\mathrm{ext}/m_s\)). But, since we are mostly interested in the limit of small \(V_p/m_s\), this contribution can be thought of as becoming arbitrary small, and we therefore don’t bother with including it in the formulas. In any case, this “conventional porewater” contribution would simply give an extra term \(-w/\rho_w\) in the equations we are about to derive, and can be included if desired.
Using eqs. 1 and 2, we get the expression for \(K_d\) in the homogeneous mixture model
\begin{equation} K_d = \frac{w\cdot\Xi }{\rho_w} + \Lambda\cdot \Xi \tag{3} \end{equation}
where we also have used the definition of the ion equilibrium coefficient \(\Xi = c^\mathrm{int}/c^\mathrm{ext}\), and utilized that \(V_p/m_s = w/\rho_w\), where \(\rho_w\) is the density of water.4
A full analysis of eq. 3 is a major task, but a few things are immediately clear:
- \(K_d\) generally has two contributions: one from Donnan equilibrium (\(w\cdot\Xi/\rho_w\)) and one from true sorption (\(\Lambda\cdot \Xi\)). Using the traditional terminology, these contributions correspond for cations to “sorption by ion exchange” and “sorption by surface complexation”, respectively. But note that eq. 3 is valid also for anions.
- For a simple cation (\(\Lambda = 0\)), \(K_d\) merely quantifies the aqueous interlayer concentration.5 As we have discussed earlier, \(K_d\) quantifies in this case a type of enhancement of the transport capacity. I think it is unfortunate that a mechanism that dominates the mass transfer capacity traditionally is labeled “sorption”.
- For cations with \(\Lambda \neq 0\), \(K_d\) is not a measure of true sorption, because we always expect a significant Donnan contribution. In this case \(K_d\) quantifies a mixture of transport enhancing and transport inhibiting mechanisms. Clearly, it is unsatisfactory to use the term “sorption” for mechanisms that both enhance and reduce transport capacity (at least when the objective is a transport description).
- For simple anions, the above expression gives a positive value for \(K_d\). Traditionally, the \(K_d\) concept has not been applied to these types of ions, and e.g. chloride is often described as “non-sorbing”, with \(K_d =0\). Since \(\Xi \rightarrow 0\) as \(w \rightarrow 0\) generally for anions, this result (\(K_d = 0\)) is recovered in this limit.6
Kd for simple cations
We end this post by examining expressions for \(K_d\) for simple cations in some specific cases. In the following we consequently assume \(\Lambda = 0\), and this section relies heavily on the ion equilibrium framework in the homogeneous mixture model, with the main relation
\begin{equation} \Xi \equiv \frac{c^\mathrm{int}}{c^\mathrm{ext}} = \Gamma f_D^{-z} \tag{4} \end{equation}
where \(z\) is the charge number of the ion, \(\Gamma \equiv \gamma^\mathrm{ext}/\gamma^\mathrm{int}\) is an activity coefficient ratio, and \(f_D = e^\frac{F\psi^\star}{RT}\) is the so-called Donnan factor, with \(\psi^\star\) (\(<0\)) being the Donnan potential.
Simple cation tracers in a 1:1 system
We assume a bentonite sample at water-to-solid mass ratio \(w\) in equilibrium with an external 1:1 solution (e.g. NaCl) of concentration \(c^\mathrm{bgr}\). The Donnan factor is in this case, in the limit \(c^\mathrm{bgr} \ll c_\mathrm{IL}\)7
\begin{equation} f_D = \Gamma_+\frac{c^\mathrm{bgr}}{c_\mathrm{IL}} \end{equation}
where \(\Gamma_+\) is the activity coefficient ratio for the cation of the 1:1 electrolyte, and, as usual
\begin{equation} c_\mathrm{IL} = \frac{CEC\cdot \rho_w}{w\cdot F} \end{equation}
where \(CEC\) is the cation exchange capacity, and \(F\) is the Faraday constant (1 eq/mol). We furthermore assume the presence of a mono-valent cation tracer, which, by definition, does not influence \(f_D\). The ion equilibrium coefficient for this tracer is (from eq. 4)
\begin{equation} \Xi = \Gamma\cdot \Omega_{1:1}\cdot \frac{\rho_w}{w} \end{equation}
where \(\Gamma\) is the activity coefficient ratio for the tracer, and we have defined
\begin{equation} \Omega_{1:1} \equiv \frac{CEC}{F\cdot c^\mathrm{bgr}\cdot\Gamma_+} \end{equation}
\(K_d\) for a simple mono-valent tracer in a 1:1 electrolyte is thus (using eq. 3 with \(\Lambda = 0\))
\begin{equation} K_{d} = \Gamma \cdot \Omega_{1:1} \tag{5} \end{equation} \begin{equation} \text{ (mono-valent simple tracer in 1:1 system)} \end{equation}
For a divalent tracer we instead have
\begin{equation} \Xi = \Gamma \cdot \Omega_{1:1}^2 \cdot \left (\frac{\rho_w}{w} \right )^2 \end{equation}
giving
\begin{equation} K_d = \Gamma \cdot \Omega_{1:1}^2 \cdot \frac{\rho_w} {w} \tag{6} \end{equation} \begin{equation}\text{(di-valent simple tracer in 1:1 system)} \end{equation}
Eqs. 5 and 6 are essentially identical8 with the expression for \(K_d\) in a 1:1 system, derived in Glaus et al. (2007), which we used in the analysis of filter influence in cation through-diffusion.
Simple cation tracers in a 2:1 system
In a 2:1 system (e.g \(\mathrm{CaCl_2}\)), the Donnan factor is, in the limit \(c^\mathrm{bgr} \ll c_\mathrm{IL}\)
\begin{equation} f_D = \sqrt{2 \Gamma_{++}\frac{c^\mathrm{bgr}}{c_\mathrm{IL}}} \end{equation}
where index “++” refers to the cation of the 2:1 background electrolyte. Thus, for a mono-valent tracer
\begin{equation} \Xi = \Gamma\cdot \sqrt{\Omega_{2:1}} \cdot \sqrt{\frac{\rho_w}{w}} \end{equation}
where
\begin{equation} \Omega_{2:1} \equiv \frac{CEC}{2F\cdot c^\mathrm{bgr}\cdot\Gamma_{++}} \end{equation}
\(K_d\) for a mono-valent simple tracer in a 2:1 electrolyte is consequently
\begin{equation} K_{d} = \Gamma \cdot \sqrt{\Omega_{2:1}}\cdot\sqrt{\frac{w}{\rho_w}} \tag{7} \end{equation} \begin{equation} \text{(simple mono-valent tracer in 2:1 system)} \end{equation}
For a divalent tracer we instead have
\begin{equation} \Xi = \Gamma \cdot \Omega_{2:1} \cdot \frac{\rho_w}{w} \end{equation}
giving
\begin{equation} K_d = \Gamma \cdot \Omega_{2:1} \tag{8} \end{equation} \begin{equation} \text{(simple di-valent tracer in 2:1 system)} \end{equation}
Density dependence of Kd
Note that \(K_d\) for a mono-valent ion in a 1:1 system does not explicitly depend on density (eq. 5), while \(K_d\) for a di-valent ion diverges as \(w\rightarrow 0\) (eq. 6). In contrast, \(K_d\) in a 2:1 system has no explicit density dependence for di-valent tracers (eq. 8), while \(K_d\) vanishes for a mono-valent tracer in the limit \(w \rightarrow 0\) (eq. 7).
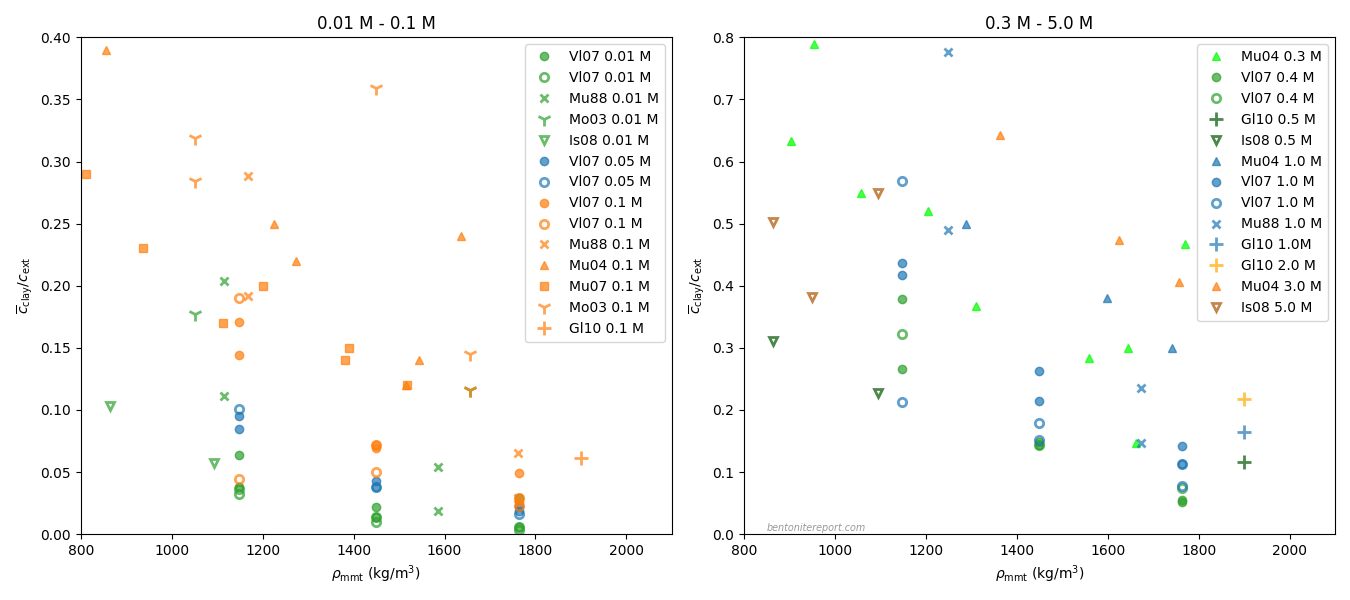
These results imply that we expect \(K_d\) to generally depend on sample density in systems where the charge number of the tracer ions differs from that of the cation of the background electrolyte. It may therefore not be appropriate to use values of \(K_d\) evaluated in batch-type tests for analyzing compacted systems.
Note also that \(K_d\) may have significant density dependence also in cases where the present analysis gives no explicit \(w\)-dependence on \(K_d\). This was demonstrated e.g. by Van Loon and Glaus (2008) for cesium tracers in sodium dominated bentonite. Interpreted in terms of the homogeneous mixture model, their results show that the interlayer activity coefficients vary significantly with density. In particular, the results imply either that the interlayer activity coefficient for cesium becomes small (\(\Gamma_\mathrm{Cs} \gg 1\)), or that the interlayer activity coefficient for sodium becomes large (\(\Gamma_\mathrm{Na} \ll 1\)), in the high density limit.
Footnotes
[1] A sediment density is, reasonably, related to e.g. initial solid-to-water ratio and to the details of the centrifugation procedure.
[2] I am not very happy with this terminology, but we need a way to distinguish this type of sorption from how the term “sorption” is used in the bentonite literature, where it nowadays essentially refers to the process of taking up an ion from a bulk water phase to some other phase. This is the reason for why there are so many quotation marks around the word “sorption” in the text.
[3] I don’t know if this is a valid assumption, but it seems like the natural starting point.
[4] The presence of water density in the formulas reflects the fact that we are using molar units (substance per unit volume), which is natural, as \(K_d\) typically has units of volume per mass. How to associate a density to water in the homogeneous mixture model is a bit subtle, and we don’t focus on that aspect here (it may be the issue of future posts). In the presented formulas \(\rho_w\) can rather be viewed as a unit conversion factor.
[5] When \(\Lambda = 0\), we can rearrange eq. 3 as
\begin{equation} \Xi = \frac{K_d\cdot \rho_w}{w} = \frac{K_d\cdot \rho_d}{\phi} \equiv \kappa \end{equation}
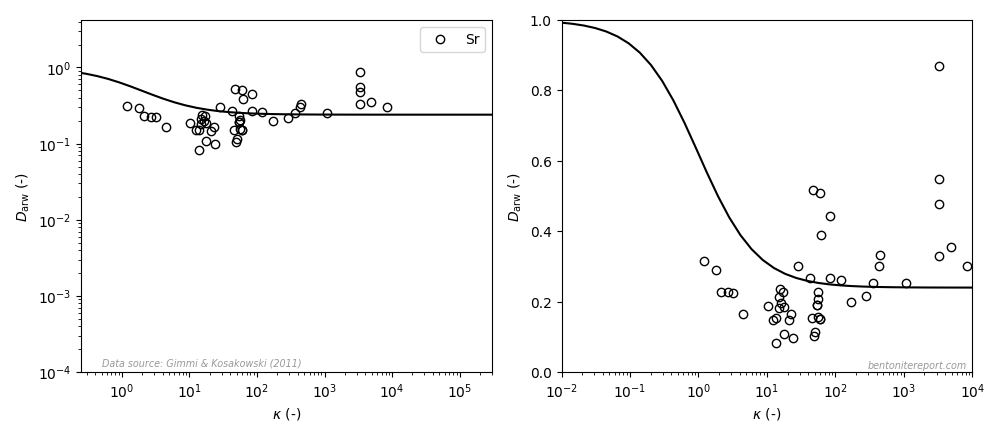
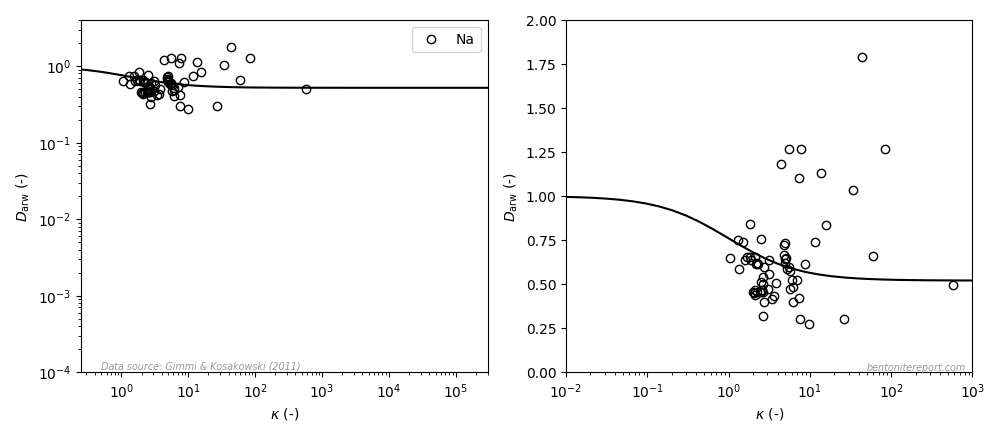
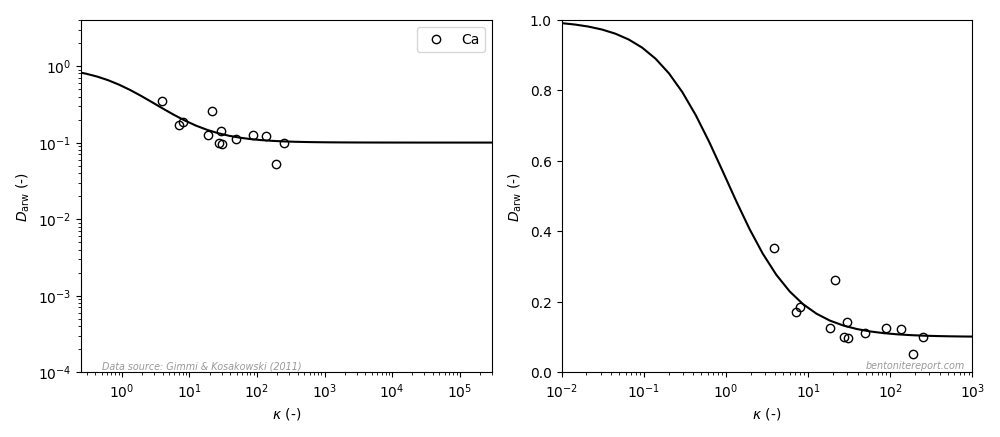
where \(\rho_d\) is dry density, \(\phi\) is porosity, and \(\kappa\) was defined as a scaled, dimensionless version of \(K_d\) by Gimmi and Kosakowski (2011), discussed in a previous blog post. Interpreted using the homogeneous mixture model, \(\kappa\) is thus simply the ion equilibrium coefficient for simple cations.
[6] By including the “conventional porewater” contribution in the definition of \(K_d\), as discussed earlier, we get for these types of anions
\begin{equation} K^\prime_d = \frac{w\cdot \Xi}{\rho_w} – \frac{w}{\rho_w} = \frac{w}{\rho_w} \left ( \Xi – 1 \right) \end{equation}
This is typically a negative quantity, and quantifies anion exclusion, in the Schofield sense of the term. We have, also with this definition, that \(K^\prime_d \rightarrow 0\) as \(w \rightarrow 0\).
[7] We assume \(c^\mathrm{bgr} \ll c_\mathrm{IL}\) in this and all following cases. For compacted bentonite \(c_\mathrm{IL}\) is of the order of several molar, and the derived approximations are thus valid for “typical” background concentrations (\(< 1\) M). Also, for an arbitrary value of \(c^\mathrm{bgr}\), one can in principle always choose a sufficiently low value of \(w\) to satisfy \(c^\mathrm{bgr} \ll c_\mathrm{IL}\).
[8] If the selectivity coefficient is identified with that derived in Birgersson (2017).