When discussing semi-permeability, we noted that a bentonite sample that is saturated with a saline solution probably contains more salt in the initial stages of the process than what is dictated by the final state Donnan equilibrium. This salt must consequently diffuse out of the sample before equilibrium is reached.
The reason for such a possible “overshoot” of the clay concentration is that an infiltrating solution is not subject to a Donnan effect (between sample and external solution) when it fills out the air-filled voids of an unsaturated sample. Also, even if the region near the interface to the external solution becomes saturated — so that a Donnan effect is active — a sample may still take up more salt than prescribed by the final state, due to hyperfiltration: with a net inflow of water and an active Donnan effect, salt will accumulate at the inlet interface (unless the interface is flushed). This increased concentration, in turn, alters the Donnan equilibrium at the interface, with the effect that more salt diffuses into the clay.
These effects are relevant for our ongoing assessment of studies of chloride equilibrium concentrations. If bentonite samples are saturated with saline solutions, without taking precautions against these effects, evaluated equilibrium concentrations may be overestimated. Note that, even if saturating a sample may be relatively fast, it may take a long time for salt to reach full equilibrium, depending on details of the experimental set-up. In particular, if the set-up is such that the external solution does not flow past the inlet, equilibration may take a very long time, being limited by diffusion in filters and tubing.
Interface excess salt
Another way for evaluated salt concentrations to overestimate the true equilibrium value — which is independent of whether or not the sample has been saturated with a saline solution — is due to excess salt at the sample interfaces.
Suppose that you determine the equilibrium salt concentration in a bentonite sample in the following way. First you prepare the sample in a test cell and contact it with an external salt solution via filters. When the system (bentonite + solution) has reached equilibrium (taking all the precautions against overestimation discussed above), the concentration profile may be conceptualized like this
The aim is to determine \(\bar{c}_\mathrm{clay}\), the
clay concentration of the species of interest
(e.g. chloride), and to relate it to the corresponding concentration in the
external solution (\(c_ \mathrm{ext}\)).
After ensuring the value of \(c_\mathrm{ext}\) (e.g. by sampling or controlling the external solution), you unload the test cell and isolate the bentonite sample. In doing so, we must keep in mind that the sample will begin to swell as soon as the force on it is released, if only water is available. In the present example it is difficult not to imagine that some water is available, e.g. in the filters.1
It is thus plausible that the actual concentration profile look
something like this directly after the sample has been isolated
We will refer to the elevated concentration at the interfaces as the
interface excess. The exact shape of the resulting
concentration profile depends reasonably on the detailed procedure for
isolating the sample.2 If the ion content of the sample is measured
as a whole, and/or if the sample is stored for an appreciable amount
of time before further analysis (so that the profile evens out due to
diffusion), it is clear that the evaluated ion content will be larger
than the actual clay concentration.
To quantify how much the clay concentration may be overestimated due
to the interface excess, we introduce an effective penetration
depth, \(\delta\)
\(\delta\) corresponds to a depth of the external concentration that
gives the same interface excess as the actual distribution. Using this
parameter, it is easy to see that the clay concentration evaluated as
the average over the entire sample is
This expression is quite interesting. We see that the relative
overestimation, reasonably, depends linearly on \(\delta\) and on the
inverse of sample length. But the expression also contains the ratio
\(r \equiv c_\mathrm{ext}/\bar{c}_\mathrm{clay}\), indicating that the effect may
be more severe for systems where the clay concentration is small in
comparison to the external concentration (high density, low
\(c_\mathrm{ext}\)).
An interface excess is more than a theoretical concept, and is frequently observed e.g. in anion through-diffusion studies. We have previously encountered them when assessing the diffusion studies of Muurinen et al. (1988) and Molera et al. (2003).3Van Loon et al. (2007) clearly demonstrate the phenomenon, as they evaluate the distribution of stable chloride (the background electrolyte) in the samples after performing the diffusion tests.4 Here is an example of the chloride distribution in a sample of density 1.6 g/cm3 and background concentration of 0.1 M5
The line labeled \(\bar{c}_\mathrm{clay}\) is evaluated from the average of only the interior sections (0.0066 M), while the line labeled \(\bar{c}_\mathrm{eval}\) is the average of all sections (0.0104 M). Using the full sample to evaluate the chloride clay concentration thus overestimates the value by a factor 1.6. From eq. 1, we see that this corresponds to \(\delta = 0.2\) mm. For a sample of length 5 mm with the same penetration depth, the corresponding overestimation is a factor of 2.1.
Here is plotted the relative overestimation (eq. 1) as a function of \(\delta\) for several systems of varying length and \(r\) (\(= c^\mathrm{ext}/\bar{c}_\mathrm{clay}\))
We see that systems with large \(r\) and/or small \(L\) become
hypersensitive to this effect. Thus, even if it may be expected that
\(\delta\) decreases with increasing \(r\)6, we may still expect an
increased overestimation for such systems.
To avoid this potential overestimation of the clay concentration, I
guess the best practice is to quickly remove the first couple of
millimeters on both sides of a sample after it has been unloaded. In
many through-diffusion tests, this is done as part of the study, as
the concentration profile across the sample often is measured. In
studies where samples are merely equilibrated with an external
solution, however, removing the interface regions may not be
considered.
Summary
We have here discussed some plausible reasons for why an evaluated
equilibrium salt concentration in a clay sample may be overestimated:
If samples are saturated directly with a saline solution. Better practice is to first saturate the sample with pure water (or a dilute solution) and then to equilibrate with respect to salt in a second stage.
If the external solution is not circulated. Diffusion may then occur over very long distances (depending on test design). The reasonable practice is to always circulate external solutions.
If interface excess is not handled. This is an issue even if saturation is done with pure water. The most convenient way to deal with this is to section off the first millimeters on both sides of the samples as quickly as possible after they are unloaded.
Footnotes
[1] One way to minimize this possible effect could be to
empty the filter before unloading the test cell. This may, however,
be difficult unless the filter itself is flushable. Also, you may
run into the problem of beginning to dry the sample.
[2] The only study I’m aware of that has
systematically investigated these types of concentration profiles is
Glaus et
al. (2011). They claim, if I understand correctly, that the
interface excess is not caused by swelling during
dismantling. Rather, they mean that the profile is the result of an
intrinsic density decrease that occurs in interface regions. Still,
they don’t discuss how swelling are supposed to be inhibited,
neither during dismantling, nor in order for the density
inhomogeneity to remain. Under any circumstance, the conclusions in
this blog post are not dependent on the cause for the presence of a
salt interface excess.
[3] In through-diffusion tests, the problem of the
interface excess is usually not that the equilibrium clay
concentration is systematically overestimated, since the detailed
concentration profile often is sampled in the final state. Instead,
the problem becomes how to separate the linear and non-linear parts
of the profile.
where \(\rho_0\) is the air density at sea level, and
\(\alpha = RT/(Mg) \approx 7500\) m is a constant. Integrating
the above formula from sea level to the height of Mount Everest
(\(\approx 9000\) m) gives
More advanced research finds a neat interpretation of this relation: the accessible height for air is 5200 m. Above this limit air is excluded, probably due to repulsion from the bedrock at these altitudes — there are reasons to believe that such rock has significantly different properties compared with rock at sea level (e.g. positive gravitational potential). In fact, both experimental work and theoretical modelling — even at the atomistic level! — have given strong evidence for the air exclusion effect: best fitting to available data is achieved with so-called air-free models.
As an example of the success of this research, one has been able to explain the existence of life in the highest regions of the Tibetan Plateu: air exists in these regions in hidden valleys (also called interpeak volumes) below the 5200 m-level, which consequently have air density \(\rho_0\). Much of present day air exclusion research is actually devoted to quantifying the amount of hidden valleys, given measurements of air density in various regions around the world (valleys that otherwise would be very difficult to discover).
Even if this research field lately has progressed heavily, there is
still a lot of exciting work waiting to be done. Of the many topics
can be mentioned so-called partial air exclusion on the outer borders
to certain high plains, air transport between hidden valleys (which
typically are connected), and the possibility of having different
accessible heights for different types of air.
A future potential application of the air exclusion effect is to build
storage e.g. for food at high altitudes. With no air around, food is
expected to stay fresh forever!
What do authors mean when they say that bentonite has semi-permeable properties? Take for example this statement, from Bradbury and Baeyens (2003)1
[…] highly compacted bentonite can function as an efficient semi-permeable membrane (Horseman et al., 1996). This implies that the re-saturation of compacted bentonite involves predominantly the movement of water molecules and not solute molecules.
Judging from the reference to Horseman et al. (1996) — which we look at below — it is relatively clear that Bradbury and Baeyens (2003) allude to the concept of salt exclusion when speaking of “semi-permeability” (although writing “solute molecules”). But a lowered equilibrium salt concentration does not automatically say that salt is less transferable.
A crucial question is what the salt is supposed to permeate. Note that
a semi-permeable component is required for defining both
swelling pressure and
salt exclusion. In case of bentonite, this component is impermeable
to the clay particles, while it is fully permeable to ions and
water (in a lab setting, it is typically a metal filter). But
Bradbury and
Baeyens (2003) seem to mean that in the process of transferring
aqueous species between an external reservoir and bentonite, salt is
somehow effectively hindered to be transferred. This does not make
much sense.
Consider e.g. the process mentioned in the quotation, i.e. to
saturate a bentonite sample with a salt solution. With
unsaturated bentonite, most bets are off regarding Donnan equilibrium,
and how salt is transferred depends on the details of the saturation
procedure; we only know that the external and internal salt
concentrations should comply with the rules for salt exclusion once
the process is finalized.
Imagine, for instance, an unsaturated sample containing bentonite
pellets on the cm-scale that very quickly is flushed with the
saturating solution, as illustrated in this state-of-the-art,
cutting-edge animation
The evolution of the salt concentration in the sample will look
something like this
Initially, as the saturating solution flushes the sample, the
concentration will be similar to that of the external concentration
(\(c_\mathrm{ext}\)). As the sample reaches saturation, it contains more
salt than what is dictated by Donnan equilibrium (\(c_\mathrm{eq.}\)),
and salt will diffuse out.
In a process like this it should be obvious that the bentonite not in any way is effectively impermeable to the salt. Note also that, although this example is somewhat extreme, the equilibrium salt concentration is probably reached “from above” in most processes where the clay is saturated with a saline solution: too much salt initially enters the sample (when a “microstructure” actually exists) and is later expelled.
Also for mass transfer between an external solution and an already saturated sample does it not make sense to speak of “semi-permeability” in the way here discussed. Consider e.g. a bentonite sample initially in equilibrium with an external 0.3 M NaCl solution, where the solution suddenly is switched to 1.0 M. Salt will then start to diffuse into the sample until a new (Donnan) equilibrium state is reached. Simultaneously (a minute amount of) water is transported out of the clay, in order for the sample to adapt to the new equilibrium pressure.2
There is nothing very “semi-permeabilic” going on here — NaCl is
obviously free to pass into the clay. That the equilibrium clay
concentration in the final state happens to be lower than in the
external concentration is irrelevant for how how difficult it is to
transfer the salt.
But it seems that many authors somehow equate “semi-permeability” with salt exclusion, and also mean that this “semi-permeability” is caused by reduced mobility for ions within the clay. E.g. Horseman et al. (1996) write (in a section entitled “Clays as semi-permeable membranes”)
[…] the net negative electrical potential between closely spaced clay particles repel anions attempting to migrate through the narrow aqueous films of a compact clay, a phenomenon known as negative adsorption or Donnan exclusion. In order to maintain electrical neutrality in the external solution, cations will tend to remain with their counter-ions and their movement through the clay will also be restricted (Fritz, 1986). The overall effect is that charged chemical species do not move readily through a compact clay and neutral water molecules may be able to pass more freely.
It must be remembered that Donnan exclusion occurs in many systemsother than “compact clay”. By instead considering e.g. a ferrocyanide solution, it becomes clear that salt exclusion has nothing to do with how hindered the ions are to move in the system (as long as they move). KCl is, of course, not excluded from a potassium ferrocyanide system because ferrocyanide repels chloride, nor does such interactions imply restricted mobility (repulsion occurs in all salt solutions). Similarly, salt is not excluded from bentonite because of repulsion between anions and surfaces (also, a negative potential does not repel anything — charge does).
In the above quotation it is easy to spot the flaw in the argument by switching roles of anions and cations; you may equally incorrectly say that cations are attracted, and that anions tag along in order to maintain charge neutrality.
The idea that “semi-permeability” (and “anion” exclusion) is
caused by mobility restrictions for the ions within the
bentonite, while water can “pass more freely” is found in many
places in the bentonite
literature. E.g. Shackelford and Moore (2013) write (where, again, potentials are
described as repelling)
In [the case of bentonite], when the clay is compressed to a sufficiently high density such that the pore spaces between adjacent clay particles are minimized to the extent that the electrostatic (diffuse double) layers surrounding the particles overlap, the overlapping negative potentials repel invading anions such that the pore becomes excluded to the anion. Cations also may be excluded to the extent that electrical neutrality in solution is required (e.g., Robinson and Stokes, 1959).
This phenomenon of anion exclusion also is responsible for the existence of semipermeable membrane behavior, which refers to the ability of a porous medium to restrict the migration of solutes, while allowing passage of the solvent (e.g., Shackelford, 2012).
[…] TOT layers bear a negative structural charge that is compensated by cation accumulation and anion depletion near their surfaces in a region known as the electrical double layer (EDL). This property gives clay materials their semipermeable membrane properties: ion transport in the clay material is hindered by electrostatic repulsion of anions from the EDL porosity, while water is freely admitted to the membrane.
and Tournassat and Steefel (2019) write (where, again, we can switch roles of “co-” and “counter-ions”, to spot one of the flaws)
The presence of overlapping diffuse layers in charged nanoporous media is responsible for a partial or total repulsion of co-ions from the porosity. In the presence of a gradient of bulk electrolyte concentration, co-ion migration through the pores is hindered, as well as the migration of their counter-ion counterparts because of the electro-neutrality constraint. This explains the salt-exclusionary properties of these materials. These properties confer these media with a semi-permeable membrane behavior: neutral aqueous species and water are freely admitted through the membrane while ions are not, giving rise to coupled transport processes.
I am quite puzzled by these statements being so commonplace.3 It does not surprise me that all the quotations basically state some version of the incorrect notion that salt exclusion is caused by electrostatic repulsion between anions and surfaces — this is, for some reason, an established “explanation” within the clay literature.4 But all quotations also state (more or less explicitly) that ions (or even “solutes”) are restricted, while water can move freely in the clay. Given that one of the main features of compacted bentonite components is to restrict water transport, with hydraulic conductivities often below 10-13 m/s, I don’t really know what to say.
Furthermore, one of the most investigated areas in bentonite research is the (relatively) high cation transport capacity that can be achieved under the right conditions. In this light, I find it peculiar to claim that bentonite generally impedes ion transport in relation to water transport.
Bentonite as a non-ideal semi-permeable membrane
As far as I see, authors seem to confuse transport between external
solutions and clay with processes that occur between two
external solutions separated by a bentonite component. Here is
an example of the latter set-up
The difference in concentration between the two solutions implies
water transport — i.e. osmosis — from the reservoir with lower salt
concentration to the reservoir with higher concentration. In this
process, the bentonite component as a whole functions as the membrane.
The bentonite component has this function because in this process it
is more permeable to water than to salt (which has a driving force to
be transported from the high concentration to the low concentration
reservoir). This is the sense in which bentonite can be said to be
semi-permeable with respect to water/salt. Note:
Salt is still transported through the bentonite. Thus, the bentonite component functions fundamentally only as a non-ideal membrane.
Zooming in on the bentonite component in the above set-up, we note that the non-ideal semi-permeable functionality emerges from the presence of two ideal semi-permeable components. As discussed above, the ideal semi-permeable components (metal filters) keep the clay particles in place.
The non-ideal semi-permeability is a consequence of salt exclusion. But these are certainly not the same thing! Rather, the implication is: Ideal semi-permeable components (impermeable to clay) \(\rightarrow\) Donnan effect \(\rightarrow\) Non-ideal semi-permeable membrane functionality (for salt)
The non-ideal functionality means that it is only relevant during non-equilibrium. E.g., a possible (osmotic) pressure increase in the right compartment in the illustration above will only last until the salt has had time to even out in the two reservoirs; left to itself, the above system will eventually end up with identical conditions in the two reservoirs. This is in contrast to the effect of an ideal membrane, where it makes sense to speak of an equilibrium osmotic pressure.
None of the above points depend critically on the membrane material being bentonite. The same principal functionality is achieved with any type of Donnan system. One could thus imagine replacing the bentonite and the metal filters with e.g. a ferrocyanide solution and appropriate ideal semi-permeable membranes. I don’t know if this particular system ever has been realized, but e.g. membranes based on polyamide rather than bentonite seems more commonplace in filtration applications (we have now opened the door to the gigantic fields of membrane and filtration technology). From this consideration it follows that “semi-permeability” cannot be attributed to anything bentonite specific (such as “overlapping double layers”, or direct interaction with charged surfaces).
I think it is important to remember that, even if bentonite is semi-permeable in the sense discussed, the transfer of any substance across a compacted bentonite sample is significantly reduced (which is why we are interested in using it e.g. for confining waste). This is true for both water and solutes (perhaps with the exception of some cations under certain conditions).
“Semi-permeability” in experiments
Even if bentonite is not semi-permeable in the sense described in many
places in the literature, its actual non-ideal semi-preamble
functionality must often be considered in compacted clay
research. Let’s have look at some relevant cases where a bentonite
sample is separated by two external solution reservoirs.
The traditional tracer through-diffusion test maintains identical
conditions in the two reservoirs (the same chemical compositions and
pressures) while adding a trace amount of the diffusing substance to
the source reservoir. The induced tracer flux is monitored by
measuring the amount of tracer entering the target reservoir.
In this case the chemical potential is identical in the two reservoirs for all components other than the tracer, and no additional transport processes are induced. Yet, it should be kept in mind that both the pressure and the electrostatic potential is different in the bentonite as compared with the reservoirs. The difference in electrostatic potential is the fundamental reason for the distinctly different diffusional behavior of cations and anions observed in these types of tests: as the background concentration is lowered, cation fluxes increase indefinitely (for constant external tracer concentration) while anion fluxes virtually vanish.
Tracer through-diffusion is often quantified using the parameter
\(D_e\), defined as the ratio between steady-state flux and
the external concentration
gradient.5 \(D_e\) is thus a
type of ion permeability coefficient, rather than a diffusion
coefficient, which it nevertheless
often is assumed to be.
Typically we have that
\(D_e^\mathrm{cation} > D_e^\mathrm{water} > D_e^\mathrm{anion}\) (where
\(D_e^\mathrm{cation}\) in principle may become
arbitrary large). This behavior both demonstrates the underlying
coupling to electrostatics, and that “charged chemical species”
under these conditions hardly can be said to move less readily through
the clay as compared with water molecules.
Measuring hydraulic conductivity
A second type of experiment where only a single component is
transported across the clay is when the reservoirs contain pure water
at different pressures. This is the typical set-up for measuring the
so-called hydraulic conductivity of a clay
component.6
Even if no other transport processes are induced (there is nothing
else present to be transported), the situation is here more complex
than for the traditional tracer through-diffusion test. The difference
in water chemical potential between the two reservoirs implies a
mechanical coupling to the clay, and a
corresponding response in density distribution. An inhomogeneous
density, in turn, implies the presence of an electric field. Water
flow through bentonite is thus fundamentally coupled to both
mechanical and electrical processes.
In analogy with \(D_e\), hydraulic conductivity is defined as the ratio
between steady-state flow and the external pressure
gradient. Consequently, hydraulic conductivity is an effective mass
transfer coefficient that don’t directly relate to the fundamental
processes in the clay.
An indication that water flow through bentonite is more subtle than what it may seem is the mere observation that the hydraulic conductivity of e.g. pure Na-montmorillonite at a porosity of 0.41 is only 8·10-15 m/s. This system thus contains more than 40% water volume-wise, but has a conductivity below that of unfractioned metamorphic and igneous rocks! At the same time, increasing the porosity by a factor 1.75 (to 0.72), the hydraulic conductivity increases by a factor of 75! (to 6·10-13 m/s7)
Mass transfer in a salt gradient
Let’s now consider the more general case with different chemical
compositions in the two reservoirs, as well as a possible pressure
difference (to begin with, we assume equal pressures).
Even with identical hydrostatic pressures in the reservoirs, this
configuration will induce a pressure response, and consequently a
density redistribution, in the bentonite. There will moreover be both
an osmotic water flow from the right to the left reservoir, as well
as a diffusive solute flux in the opposite direction. This general
configuration thus necessarily couples hydraulic, mechanical,
electrical, and chemical processes.
This type of configuration is considered e.g. in the study of osmotic effects in geological settings, where a clay or shale formation may act as a membrane.8 But although this configuration is highly relevant for engineered clay barrier systems, I cannot think of very many studies focused on these couplings (perhaps I should look better).
For example, most through-diffusion studies are of the tracer type discussed above, although evaluated parameters are often used in models with more general configurations (e.g. with salt or pressure gradients). Also, I am not aware of any measurements of hydraulic conductivity in case of a salt gradient (but the same hydrostatic pressure), and I am even less aware of such values being compared with those evaluated in conventional tests (discussed previously).
A quite spectacular demonstration that mass transfer may occur very differently in this general configuration is the seeming steady-stateuphill diffusion effect: adding an equal concentration of a cation tracer to the reservoirs in a set-up with a maintained difference in background concentration, a tracer concentration difference spontaneously develops. \(D_e\) for the tracer can thus equal infinity,9 or be negative (definitely proving that this parameter is not a diffusion coefficient). I leave it as an exercise to the reader to work out how “semi-permeable” the clay is in this case. Update (240822):The “uphill” diffusion effect is further discussed here.
A process of practical importance for engineered clay barrier systems
is hyperfiltration of salts. This process will occur when a sufficient
pressure difference is applied over a bentonite sample contacted with
saline solutions. Water and salt will then be transferred in the same
direction, but, due to exclusion, salt will accumulate on the
inlet side. A steady-state concentration profile for such a process
may look like this
The local salt concentration at the sample interface on the inlet side
may thus be larger than the concentration of the injected
solution. This may have consequences e.g. when evaluating hydraulic
conductivity using saline solutions.
Hyperfiltration may also influence the way a sample becomes saturated, if saturated with a saline solution. If the region near the inlet is virtually saturated, while regions farther into the sample still are unsaturated, hyperfiltration could occur. In such a scenario the clay could in a sense be said to be semi-permeable (letting through water and filtrating salts), but note that the net effect is to transfer more salt into the sample than what is dictated by Donnan equilibrium with the injected solution (which has concentration \(c_1\), if we stick with the figure above). Salt will then have to diffuse out again, in later stages of the process, before full equilibrium is reached. This is in similarity with the saturation process that we considered earlier.
[2] This is more than a thought-experiment; a test just like this was conducted by Karnland et al. (2005). Here is the recorded pressure response of a Na-montmorillonite sample (dry density 1.4 g/cm3) as it is contacted with NaCl solutions of increasing concentration
[3] As a side note, is the region near the surface supposed to be called “diffuse layer”, “electrical double layer”, or “electrostatic (diffuse double) layer”?
[5] This is not a gradient in the mathematical sense, but is defined as \( \left (c_\mathrm{target} – c_\mathrm{source} \right)/L\), where \(L\) is sample length.
[6] Hydraulic conductivity is often also measured
using a saline solution, which is commented on below.
[7] Which
still is an a amazingly small hydraulic conductivity, considering
the the water content.
[9] Mathematically, the statement “equal infinity” is
mostly nonsense, but I am trying to convey that a there is a tracer
flux even without any external tracer concentration difference.
Mo03 performed both chloride and iodide through-diffusion tests on
“MX-80” bentonite, but here we focus on the chloride
results. However, since the only example in the paper of an outflux
evolution and corresponding concentration profile is for iodide, this
particular result will also be investigated. The tests were performed
at background concentrations of 0.01 M or 0.1 M NaClO4, and nominal
sample densities of 0.4, 0.8, 1.2, 1.6, and 1.8 g/cm3. We refer to a
single test by stating “nominal density/background concentration”,
e.g. a test performed at nominal density 1.6 and background
concentration 0.1 M is referred to as “1.6/0.1”.
Uncertainty of samples
The material used is discussed only briefly, and the only reference given for its properties is (Müller-Von Moos and Kahr, 1983). I don’t find any reason to believe that the “MX-80” batch used in this study actually is the one investigated in this reference, and have to assume the same type of uncertainty regarding the material as we did in the assessment of Muurinen et al (1988). I therefore refer to that blog post for a discussion on uncertainty in montmorillonite content, cation population, and soluble calcium minerals.
Density
The samples in Mo03 are cylindrical with radius 0.5 cm and length 0.5
cm, giving a volume of 0.39 cm3. This is quite small, and corresponds
e.g. only to about 4% of the sample size used in
Muurinen et al
(1988). With such a small volume, the samples are at the
limit for being considered as a homogeneous material, especially for
the lowest densities: the samples of density 0.4 g/cm3 contain 0.157 g
dry substance in total, while a single 1 mm3 accessory grain weighs
about 0.002 — 0.003 g.
Furthermore, as the samples are sectioned after termination, the
amount substance in each piece may be very small. This could cause
additional problems, e.g. enhancing the effect of drying. The
reported profile (1.6/0.1, iodide diffusion) has 10 sections in the
first 2 mm. As the total mass dry substance in this sample is 0.628 g,
these sections have about 0.025 g dry substance each (corresponding to
the mass of about ten 1 mm3 grains). For the lowest density, a similar
sectioning corresponds to slices of dry mass 0.006 g (the paper does
not give any information on how the low density samples were
sectioned).
Mo03 only report nominal densities for the samples, but from the above considerations it is clear that a substantial (but unknown) variation may be expected in densities and concentrations.
A common feature of many through-diffusion studies is that the sample
density appears to decrease in the first few millimeters near the
confining filters. We saw this effect in the profiles of
Muurinen et al (1988),
and it has been the topic of some
studies,
including Mo03. Here, we don’t consider any possible cause, but simply
note that the samples seem to show this feature quite generally (below
we discuss how Mo03 handle this). Since the samples of Mo03 are only
of length 5 mm, we may expect that the major part of them are affected
by this effect. Of course, this increases the uncertainty of the
actual density of the used samples.
Uncertainty of external solutions
Mo03 do not describe how the external solutions were prepared, other
than that they used high grade chemicals. We assume here that the
preparation did not introduce any significant uncertainty.
Since “MX-80” contains a substantial amount of divalent ions, connecting this material with (initially) pure sodium solutions inevitably initiates cation exchange processes. The extent of this exchange depends on details such as solution concentrations, reservoir volumes, number of solution replacements, time, etc…
Very little information is given on the volume of the external solution
reservoirs. It is only hinted that the outlet reservoir may be 25 ml,
and for the inlet reservoir the only information is
The volume of the inlet reservoir was sufficient to keep the
concentration nearly constant (within a few percent) throughout
the experiments.
Consequently, we do not have enough information to assess the exact ion population during the course of the tests. We can, however, simulate this process of “unintentional exchange” to get some appreciation for the amount of divalent ions still left in the sample, as we did in the assessment of Muurinen et al. (1988). Here are the results from calculating the exchange equilibrium between a sample initially containing 30% exchangeable charge in form of calcium (70% sodium), and external NaClO4 solutions of various concentrations and volumes
In these calculations we assume a sample of density 1.6 g/cm3 (except
when indicated), a volume of 0.39 cm3, a cation exchange capacity of
0.75 eq/kg, and a Ca/Na selectivity coefficient of 5.
These simulations make it clear that the tests performed at 0.01 M
most probably contain most of the divalent ions initially present in
the “MX-80” material: even with an external solution volume of 1000
ml, or with density 0.4 g/cm3, exchange is quite
limited. For the tests performed at 0.1 M we expect some exchange of
the divalent ions, but we really can’t tell to what extent, as the
exact value strongly depends on handling (solution volumes, if
solutions were replaced, etc.). That the exact ion population is
unknown, and that the divalent/monovalent ratio probably is different
for different samples, are obviously major problems of the study (the
same problems were identified
in Muurinen et al
(1988)).
Uncertainty of diffusion parameters
Diffusion model
Mo03 determine diffusion parameters by fitting a model to all
available data, i.e the outflux evolution and the concentration
profile across the sample at termination. The model is solved by a
numerical code (“ANADIFF”) that takes into account transport both in
clay samples and filters. The fitted parameters are an apparent
diffusivity, \(D_a\), and a so-called “capacity factor”,
\(\alpha\). \(\alpha\) is vaguely interpreted as being the combination of
a porosity factor \(\epsilon\), and a sorption distribution
coefficient \(K_d\), described as “a generic term devoid of mechanism”
It is claimed that for anions, \(K_d\) can be treated as negative, giving \(\alpha < \epsilon\). I have criticized this mixing of what actually are incompatible models in an earlier blog post. Strictly, this use of a “generic term devoid of mechanism” means that the evaluated \(\alpha\) should not be interpreted in any particular way. Nevertheless, the waythis study is referenced in otherpublications, \(\alpha\) is interpreted as an effective porosity. It should be noticed, however, that this study is performed with a background electrolyte of NaClO4. The only chloride (or iodide) present is therefore at trace level, and it cannot be excluded that a mechanism of true sorption influences the results (there are indications that this is the case in other studies).
For the present assessment we anyway assume that \(\alpha\) directly
quantifies the anion equilibrium between clay and the external
solution (i.e. equivalent to
the
incorrect way of
assuming that \(\alpha\) quantifies a volume accessible to
chloride). It should be kept in mind, though, that effects of anion
equilibrium and potential true sorption is not resolved by the
single parameter \(\alpha\).
where \(c\) is the concentration in the clay of the isotope under
consideration, and the diffusion coefficient is written \(D_p\) to
acknowledge that it is a pore diffusivity (when referring to models
and parameter evaluations in Mo03 we will use the notation
“\(D_a\)”). The boundary conditions are
Oddly, Mo03 model the system as if two independent diffusion processes are simultaneously active. They refer to these as the “fast” and the “slow” processes, and hypothesize that they relate to diffusion in interlayer water2 and “interparticle water”,3 respectively.
The “fast” process is the “ordinary” process that is assumed to reach steady state during the course of the test, and that is the focus of other through-diffusion studies. The “slow” process, on the other hand, is introduced to account for the frequent observation that measured tracer profiles are usually significantly non-linear near the interface to the source reservoir (discussed briefly above). I guess that the reason for this concentration variation is due to swelling when the sample is unloaded. But even if the reason is not fully clear, it can be directly ruled out that it is the effect of a second, independent, diffusion process — because this is not how diffusion works!
If anions move both in interlayers and “interparticle water”, they reasonably transfer back and forth between these domains, resulting in a single diffusion process (the diffusivity of such a process depends on the diffusivity of the individual domains and their geometrical configuration). To instead treat diffusion in each domain as independent means that these processes are assumed to occur without transfer between the domains, i.e. that the bentonite is supposed to contain isolated “interlayer pipes”, and “interparticle pipes”, that don’t interact. It should be obvious that this is not a reasonable assumption. Incidentally, this is how all multi-porous models assume diffusion to occur (while simultaneously assuming that the domains are in local equilibrium…).
We will thus focus on the “fast” process in this assessment, although we also use the information provided by the parameters for the “slow” process. Mo03 report the fitted values for \(D_a\) and \(\alpha\) in a table (and diagrams), and only show a comparison between model and measured data in a single case: for iodide diffusion at 0.1 M background concentration and density 1.6 g/cm3. To make any kind of assessment of the quality of these estimations we therefore have to focus on this experiment (the article states that these results are “typical high clay density data”).
Outflux
The first thing to note is that the modeled accumulated diffusive substance does not correspond to the analytical solution for the diffusion process. Here is a figure of the experimental data and the reported model (as presented in the article), that also include the solution to eqs. 1 and 2.
In fact, the model presented in Mo03 has an incorrect time dependency in the early stages. Here is a comparison between the presented model and analytical solutions in the transient stage
With the given boundary conditions, the solutions to the diffusion
equation inevitably has zero slope at \(t = 0\),4 reflecting
that it takes a finite amount of time for any substance to reach the
outflux boundary. The models presented in Mo03, on the other hand, has
a non-zero slope in this limit. I cannot understand the reason for
this (is it an underlying problem with the model, or just a graphical
error?), but it certainly puts all reported parameter values in doubt.
The preferred way to evaluate diffusion data is, in my opinion, to look
at the flux evolution rather than the evolution of the accumulated
amount of diffused substance. Converting the reported data to flux,
gives the following picture.5
From a flux evolution it is easier to establish the steady-state, as it reaches a constant. It furthermore gives a better understanding for how well constrained the model is by the data. As is seen from the figure, the model is not at all very well constrained, as the experimental data almost completely miss the transient stage. (And, again, it is seen that the model in the paper with \(D_a= 9\cdot 10^{-11}\) m/s2 does not correspond to the analytical solution.)
The short transient stage is a consequence of using thin samples (0.5 cm). Compared e.g. to Muurinen et al (1988), who used three times as long samples, the breakthrough time is here expected to be \(3^2 = 9\) times shorter. As Muurinen et al. (1988) evaluated breakthrough times in the range 1 — 9 days, we here expect very short times. Here are the breakthrough times for all chloride diffusion tests, evaluated from the reported diffusion coefficients (“fast” process) using the formula \(t_\mathrm{bt} = L^2/(6D_a)\).
Test
\(D_a\)
\(t_\mathrm{bt}\)
(m2/s)
(days)
0.4/0.01
\(8\cdot 10^{-10}\)
0.06
0.4/0.1
\(9\cdot 10^{-10}\)
0.05
0.4/0.1
\(8\cdot 10^{-10}\)
0.06
0.8/0.01
\(3.5\cdot 10^{-10}\)
0.14
0.8/0.1
\(3.5\cdot 10^{-10}\)
0.14
0.8/0.1
\(3.7\cdot 10^{-10}\)
0.13
1.2/0.01
\(1.4\cdot 10^{-10}\)
0.34
1.2/0.1
\(2.3\cdot 10^{-10}\)
0.21
1.2/0.1
\(2.0\cdot 10^{-10}\)
0.24
1.6/0.1
\(1.0\cdot 10^{-10}\)
0.48
1.8/0.01
\(2\cdot 10^{-11}\)
2.41
1.8/0.1
\(5\cdot 10^{-11}\)
0.96
1.8/0.1
\(5.5\cdot 10^{-11}\)
0.88
The breakthrough time is much shorter than a day in almost all tests! To sample the transient stage properly requires a sampling frequency higher than \(1/t_{bt}\). As seen from the provided example of a outflux evolution, this is not the case: The second measurement is done after about 1 day, while the breakthrough time is about 0.5 days (moreover, the first measurement appears as an outlier). We have no information on sampling frequency in the other tests, but note that to properly sample e.g. the tests at 0.8 g/cm3 requires measurements at least every third hour or so. For 0.4 g/cm3, the required sample frequency is once an hour! This design choice puts more doubt on the quality of the evaluated parameters.
Concentration profile
The measured concentration profile across the 1.6/0.1 iodide sample,
and corresponding model results are presented in Mo03 in a figure very
similar to this
Here the two models correspond to the “slow” and “fast” process discussed above (a division, remember, that don’t make sense). Zooming in on the “linear” part of the profile, we can compare the “fast” process with analytical solutions (eqs. 1 and 2)
The analytical solutions correspond directly to the outflux curves presented above. We note that the analytical solution with \(D_p = 9\cdot 10^{-11}\) m/s2 corresponds almost exactly to the model presented by Mo03. As this model basically has the same steady state flux and diffusion coefficient, we expect this similarity. It is, however, still a bit surprising, since the corresponding outflux curve of the model in Mo03 was seen to not correspond to the analytical solution. This continues to cast doubt on the model used for evaluating the parameters.
We furthermore note that the evolution of the activity of the source
reservoir is not reported. Once in the text is mentioned that the
“carrier concentration” is \(10^{-6}\) M, but since we don’t know how
much of this concentration corresponds to the radioactive isotope, we
can not directly compare with reported concentration profile across
the sample (whose concentration unit is counts per minute per cm3).
By extrapolating the above model curve with \(\alpha = 0.15\), we can
however deduce that the corresponding source activity for this
particular sample is \(C_0 = 1.26\cdot 10^5/0.15\) cpu/cm3
\(= 8.40\cdot 10^5\) cpu/cm3. But it is unsatisfying that we cannot
check this independently. Also, we can of course not assume that this
value of \(C_0\) is the same in any other of the tests (in particular
those involving chloride). We thus lack vital information (\(C_0\)) to
be able to make a full assessment of the model fitting.
It should furthermore be noticed that the experimental concentration
profile does not constrain the models very well. Indeed, the adopted
model (diffusivity \(9\cdot 10^{-11}\) m/s2) misses the two
rightmost concentration points (which corresponds to half the
sample!). A model that fits this part of the profile has a
considerable higher diffusivity, and a correspondingly lower
\(\alpha\) (note that the product \(D_p\cdot \alpha\) is constrained
by the steady-state flux, eq. 3).
More peculiarities of the modeling is found if looking at the “slow”
process (remember that this is not a real diffusion process!). Zooming
in on the interface part of the profile and comparing with analytical
solutions gives this picture
Here we note that an analytical solution coincides with the model presented in Mo03 with parameters \(D_a = 6\cdot 10^{-14}\) m2/s and \(\alpha = 1.12\) only if it is propagated for about 15 days! Given that no outflux measurements seem to have been performed after about 4 days (see above), I don’t now what to make of this. Was the test actually conducted for 15 days? If so, why is not more of the outflux measured/reported? (And why were the samples then designed to give a breakthrough time of only a few hours?)
Without knowledge of for how long the tests were conducted, the reported diffusion parameters becomes rather arbitrary, especially for the low density samples. For e.g. the samples of density 0.4 g/cm3, even the “slow” process has a diffusivity high enough to reach steady-state within a few days. Simulating the processes with the reported parameters gives the following profiles if evaluated after 1 and 4 days, respectively
The line denoted “total” is what should resemble the measured
(unreported) data. It should be clear from these plots that the
division of the profile into two separate parts is quite arbitrary. It
follows that the evaluated diffusion parameters for the process of
which we are interested (“fast”) has little value.
Summary and verdict
We have seen that the reported model fitting leaves a lot of unanswered questions: some of the model curves don’t correspond to the analytical solutions, information on evolution times and source concentrations is missing, and the modeled profiles are divided quite arbitrary into two separate contributions (which are not two independent diffusion process).
Moreover, the ion population (divalent vs. monovalent cations) of the samples are not known, but there are strong reasons to believe that the 0.01 M tests contain a significant amount of divalent ions, while the 0.1 M samples are partly converted to a more pure sodium state.
Also, the small size of the samples contributes to more uncertainty,
both in terms of density, but also for the flux evolution because the
breakthrough times becomes very short.
Based on all of these uncertainties, I mean that the results of Mo03
does not contribute to quantitative process understanding and my
decision is to not to use the study for e.g. validating models
of anion exclusion.
A confirmation of the uncertainty in this study is given by
considering the density dependence on the chloride equilibrium
concentrations for constant background concentration, evaluated from
the reported diffusion parameters (\(\alpha\) for the “fast” process).
If these results should be taken at face value, we have to accept a
very intricate density dependence: for 0.1 M background, the
equilibrium concentration is mainly constant between densities 0.3
g/cm3 and 0.7 g/cm3, and increases
between densities 1.0 g/cm3 and 1.45 g/cm3 (or,
at least, does not decrease). For 0.01 M background, the equilibrium
concentration instead falls quite dramatically between between
densities 0.3 g/cm3 and 0.7 g/cm3, and
thereafter displays only a minor density dependence.
To accept such dependencies, I require a considerably more rigorous experimental procedure and evaluation. In this case, I rather view the above plot as a confirmation of large uncertainties in parameter evaluation and sample properties.
[1] Strictly, \(c(0,t)\) relates to the concentration in the endpoint of the inlet filter. But we ignore filter resistance in this assessment, which is valid for the 1.6/0.1 sample. Moreover, the filter diffusivities are not reported in Mo03.
[2] Mo03 refer to interlayer pores as
“intralayer” pores, which may cause some confusion.
[3] Apparently, the authors assume an
underlying
stack view of the material.
[4] It may be
objected that the analytical solution do not include the filter
resistance. But note that filter resistance only will increase the
delay. Moreover, the transport capacity of the sample in this test
is so low that filters have no significant influence.
[5] The model by Mo03 looks noisy
because I have read off values of accumulated concentration from the
published graph. The “noise” occurs because the flux is evaluated
from the concentration data by the difference formula:
where \(t_i\) and \(t_{i+1}\) are the time coordinates for two consequitive data points, \(a(t)\) is the accumulated amount diffused substance at time \(t\), \(A\) is the cross sectional area of the sample, \(\bar{t}_i = (t_{i+1} + t_i)/2\) is the average time of the considered time interval, and \(\bar{j}\) denotes the average flux during this time interval.
We havediscussedvariousaspects of “anionexclusion” on this blog. This concept is often used to justify multi-porosity models of compacted bentonite, by reasoning that the exclusion mechanism makes parts of the pore space inaccessible to anions. But we have seen that this reasoning has no theoretical backup: studies making such assumptions usually turn out to refer to conventional electric double layer theory, described e.g. by the Poisson-Boltzmann equation. In the following, we refer to the notion of compartments inaccessible to anions as complete anion exclusion.
In fact, a single, physically reasonable concept underlies basically all descriptions of anion exclusion in the clay literature: charge separation. Although the required mathematics may differ for different systems — may it be using Donnan’s “classical equations”, or the Poisson-Boltzmann equation — the underlying mechanism is the same. In the following we refer to this type of description as traditional theory or Donnan theory. It is important to recognize that traditional theory is incompatible with complete anion exclusion: the Poisson-Boltzmann equation predicts anions everywhere.
In more recent years, however, a different meaning of the term “anion
exclusion” has sneaked into the literature. This seems to be related
to the dawn of molecular dynamics (MD) simulations of clays. In
particular, the study of Rotenberg et al. (2007) — which I think is the first published MD
simulation of montmorillonite interlayers in contact with an external
compartment — is frequently cited as demonstrating qualitatively
different results as compared with the traditional
models. E.g. Kosakowski and Berner (2013) write
Very often it is assumed that negatively charged ions are strongly hindered to enter the interlayer space (Kosakowski et al., 2008; Rotenberg et al., 2007), although other authors come to different conclusions (Karnland et al., 2007). Note that we favor the former view with our montmorillonite setup.
Although the terms “assumed” and “conclusions” seem misplaced, it
is clear that Kosakowski and Berner (2013) mean that the interlayer space is
essentially anion-free, rather than obeying ordinary Donnan
equilibrium (the approach used in
Karnland et
al. (2007)).
The interlayer space can be seen as an extreme case where the diffuse layer vanishes leaving only the Stern layer of the adjacent basal surfaces. For this reason, the interlayer space is often considered to be completely free of anions (Tournassat and Appelo 2011), although this hypothesis is still controversial (Rotenberg et al. 2007c; Birgersson and Karnland 2009).
Based upon [results from anion diffusion tests], anion-exclusion models have been formulated, which subdivide the water-filled pore space into interlayer, diffuse (or electric) double layer (DDL) and “free” water porosities (Wersin et al. 2004; Tournassat & Appelo 2011; Appelo 2013). In this formulation, anions are considered to reside in the “free” electrically neutral solution and in the DDL in the external (intergranular) pores, whereas the interlayer (intragranular) space is considered devoid of anions. Support for this model has been given by molecular dynamics simulations (Rotenberg et al. 2007), but this issue remains controversial (Birgersson & Karnland 2009)
The term “anion-exclusion” is here fully transformed to refer to complete exclusion, rather than to the traditional theory from which the term was coined. Note that the picture of bentonite given in this and the previous quotations is basically the contemporary mainstream view, which we discussed in a previous blog post. This description has not emerged from considering MD results that are allegedly in contradiction with traditional Donnan equilibrium theory. Rather, it has resulted from misusing the concept of exclusion-volume. The study of Rotenberg et al. (2007) (Rot07, in the following) supports the contemporary mainstream view only to the extent that it is at odds with the predictions of traditional theory. But is it? Let’s take a look at the relevant MD studies.
Rotenberg et al. (2007)
Rot07 is not primarily a study of the anion equilibrium, but considers more generally the transition of species between an external compartment2 and interlayer pores: water, cations (Na and Cs), and anions (Cl). The study only concerns interlayers with two monolayers of water, in the following referred to as a 2WL system. There is of course nothing wrong with exclusively studying the 2WL system, but this study alone cannot be used to support general model assumptions regarding interlayers (which anyway is commonplace, as we saw above). The meaning of the term “interlayer” in modern clay literature is quite confusing, but there is at least full consensus that it includes also states with three monolayers of water (3WL) (we’ll get back to those). Rot07 furthermore consider only a single external concentration, of 0.52 M.
Here is an illustration of the simulated system:
A cell (outlined with dashed lines) containing two montmorillonite
layers (yellow) and six chloride ions (green) is repeated infinitely
in all directions (the cell depth in the direction normal to the
picture is 20.72 Å). While only chloride ions are indicated in this
figure, also cations, water atoms, and montmorillonite atoms are
explicitly accounted for in the simulation.
Note that the study neither varies density (interlayer distance) nor
external concentration (number of chloride ions) — two variables
essential for studying anion equilibrium. I don’t mean this as direct
criticism, but it should be recognized when the study is used to
support assumptions regarding interlayers in other models.
What I do want to criticize, however, is that
Rot07 don’t
actually compare with Donnan theory. Instead, they seem to be under
the impression that traditional theory predicts complete exclusion in
their system. Consider this passage in the introduction
Due to the negative charge of clay layers, anions should be repelled by the external surfaces, and excluded from the interlayers. On the contrary, cations are attracted by the surfaces, and may exchange with the natural interlayer counterions.
Here they associate two different terms with the anions: they are
repelled by the “external surfaces” and excluded from
“interlayers”.
I can only interpret this as meaning that anions are completely
excluded from interlayers, especially as the wording “on the
contrary” is used when describing cations.3
The study comprises both a “plain” MD simulation of the (presumed) equilibrium state, and separate calculations of free energy profiles. In the “plain” MD simulation, anions do not enter the interlayers, and the calculation of the free energy profile gives a barrier of ~9 kT for chloride to enter the interlayer.
These results motivate the authors to conclude that the “thermal fluctuations do not allow anions to overcome the free energy barrier corresponding to their entrance into the interlayer” and that “anions are excluded from the interlayer: the probability for an anion reaching the interface to enter into the interlayer is very small (of the order of e-9 ~ 10-4)”
It is important to keep in mind that the authors are under the
impression that this result and conclusion are in line with the
traditional description of anion
exclusion.3 When summarizing
their findings they write
All the results are in agreement with the common sense on ionic exchange and anion exclusion.
and
The results confirm the generally admitted ideas of ionic exchange and anion exclusion
The problem is that this “common sense” and these “generally
admitted ideas” are based on
misconceptions of traditional theory (I also think one should be
careful with using terms like these in scientific
writing). Consequently, the authors erroneously conclude that their
results confirm, rather than contrast, traditional theory. This is
opposite to how this study is referred to in later publications, as
was exemplified above.
The anion exclusion predicted from Donnan theory for the system in
Rot07 is estimated
as follows. The adopted montmorillonite unit cell
(Na0.75Si8Al3.25Mg0.75O20OH4)
has
structural charge 0.75e, and lateral dimensions 8.97 Å × 5.18
Å. With an interlayer width of 6.1 Å we thus have for the
concentration of interlayer charge
where \(N_A\) is the
Avogadro
constant. Using this value for \(c_{IL}\) in the expression for
internal anion concentration in an ideal
1:1 Donnan system,
This should be the anion interlayer concentration expected from “generally admitted ideas”, and Rot07 should have concluded that their results differ by a factor ~1000 (or more) from traditional theory. This is not to say that the calculations are incorrect (more on that later), but it certainly puts the results in a different light. A discrepancy of this magnitude should reasonably be of interest to investigate further.
Hsiao and Hedström (2015)
Considerably more detailed MD simulations of the 2WL system are
provided by Hsiao and
Hedström (2015) (Hsi15, hereafter). In contrast to
Rot07,
Hsi15 specifically
focus on the anion equilibrium, and they explicitly compare with both
conventional Donnan theory, and the results of
Rot07. In these
simulations, chloride actually populates the interlayer.
Hsi15 also analyze the convergence behavior, by varying system size and simulation time. This analysis makes it clear both that most of the simulations presented in the paper are properly converged, and that the simulation of Rot07 is not. With external concentration 1.67 M, Hsi15 demonstrate that, during intervals of 20 ns, the interlayer concentration fluctuates between basically zero and 0.13 M (converged value: 0.04 M), in a system with similar size as that of Rot07. Given that the total simulation time of the earlier study is 20 ns, and that it also adopts a considerably lower external concentration, its result of zero chloride concentration in the interlayer is no surprise.
The converged interlayer concentrations in
Hsi15 look like
this in the direction normal to the basal surfaces (simulation time:
150 ns, layer size: 8 × 4 unit cells, external concentration:
1.67 M)
Note that the simulation contains two interlayer pores (indicated by the dotted lines; cf. the illustration of the simulated system) and that sodium and chloride populate the same central layer, sandwiched by the two water layers (not shown). The nearly identical chloride profiles is a strong confirmation that the simulation is converged.
The chloride interlayer concentrations evaluated in Hsi15 deviate strongly from the predictions of the ideal Donnan formula. With \(c_{IL}\) = 4.23 M (as reported in the article) and \(c^\mathrm{ext}\) = 1.67 M, eq. 1 gives \(c^\mathrm{int}\) = 0.580 M, while the MD results are in the range 0.033 M — 0.045 M, i.e. more than a factor 10 lower (but not a factor 1000).
Hsi15 also calculate
the free energy profiles along the coordinate connecting the external
compartment and the interlayer, similar to the technique utilized by
Rot07 (as far as I
understand). For the external concentration of 1.67 M they evaluate a
free energy barrier of ~3.84 kT, which corresponds to an
interlayer concentration of 0.036 M, and is in good agreement with the
directly evaluated concentrations.
Note that Hsi15 —
in contrast to Rot07 — conclude significant deviation between the MD results of
the 2WL system and ideal traditional theory. Continuing their
investigation (again, in contrast to
Rot07),
Hsi15 found that the
contribution from ion hydration to the free energy barrier basically
make up for the entire discrepancy with the ideal Donnan formula.
Moreover, even though the ideal Donnan formula strongly overestimates
the actual values obtained from MD, it still shows the correct
dependency on external concentration: when the external
concentration is lowered to 0.55 M, the evaluated free energy barrier
increases to ~5.16 kT, which corresponds to a reduction of the
internal concentration by about a factor of 10. This is in
agreement with Donnan theory, which gives for the expected
reduction (0.55/1.67)2 ≈ 0.11.
From the results of Hsi15 (and Rot07,
for that matter), a relatively clear picture emerges: MD simulated 2WL
systems function as Donnan systems. Anions are not completely
excluded, and the dependency on external concentration is in line with
what we expect from
a varying Donnan potential across the interface between interlayer
and external compartment
(Hsi15 even comment
on observing the space-charge region!).
The simulated 2WL system is, however, strongly non-ideal, as a consequence of the ions not being optimally hydrated. Hsi15 remark that the simulations probably overestimate this energy cost, e.g. because atoms are treated as non-polarizable. This warning should certainly be seriously considered before using the results of MD simulated 2WL systems to motivate multi-porosity in compacted bentonite. But, concerning assumptions of complete anion exclusion in interlayers, another system must obviously also be considered: 3WL.
Hedström and Karnland (2012)
MD simulations of anion equilibrium in the 3WL system are presented in
Hedström and
Karnland (2012) (Hed12, in the following).
Hed12 consider
three different external concentrations, by including either 12, 6, or
4 pairs of excess ions (Cl– + Na+). This study
also varies the way the interlayer charge is distributed, by either
locating unit charges on specific magnesium atoms in the
montmorillonite structure, or by evenly reducing the charge by a minor
amount on all the octahedrally coordinated atoms.
Here are the resulting ion concentration profiles across the
interlayer, for the simulation containing 12 chloride ions, and evenly
distributed interlayer charge (simulation time: 20 ns, layer size:
4 × 4 unit cells)
Chloride mainly resides in the middle of the interlayer also in the 3WL system, but is now separated from sodium, which forms two off-center main layers. The dotted lines indicate the extension of the interlayer.
The main objectives of this study are to simply establish that anions in MD equilibrium simulations do populate interlayers, and to discuss the influence of unavoidable finite-size effects (6 and 12 are, after all, quite far from Avogadro’s number). In doing so, Hed12 demonstrate that the system obeys the principles of Donnan equilibrium, and behaves approximately in accordance with the ideal Donnan formula (eq. 1). The authors acknowledge, however, that full quantitative comparison with Donnan theory would require better convergence of the simulations (the convergence analysis was further developed in Hsi15). If we anyway make such a comparison, it looks like this
#Cl TOT
Layer charge
#Cl IL
\(c^\mathrm{ext}\)
\(c^\mathrm{int}\) (Donnan)
\(c^\mathrm{int}\) (MD)
12
distr.
1.8
1.45
0.62
0.42 (67%)
12
loc.
1.4
1.50
0.66
0.32 (49%)
6
distr.
0.6
0.77
0.20
0.14 (70%)
6
loc.
1.3
0.67
0.15
0.30 (197%)
4
distr.
0.2
0.54
0.10
0.05 (46%)
4
loc.
0.18
0.54
0.10
0.04 (41%)
The first column lists the total number of chloride ions in the simulations, and the second indicates if the layer charge was distributed on all octahedrally coordinated atoms (“distr.”) or localized on specific atoms (“loc.”) The third column lists the average number of chloride ions found in the interlayer in each simulation. \(c^\mathrm{ext}\) denotes the corresponding average molar concentration in the external compartment. The last two columns lists the corresponding average interlayer concentration as evaluated either from the Donnan formula (eq. 1 with \(c_{IL}\) = 2.77 M, and the listed \(c^\mathrm{ext}\)), or from the simulation itself.
The simulated results are indeed within about a factor of 2 from the predictions of ideal Donnan theory, but they also show a certain variation in systems with the same number of total chloride ions,4 indicating incomplete convergence (compare with the fully converged result of Hsi15). It is also clear from the analysis in Hed12 and Hsi15 that the simulations with the highest number och chloride ions (12) are closer to being fully converged.5 Let’s therefore use the result of those simulations to compare with experimental data.
Comparison with experiments
In an earlier blog post, we looked at the available experimental data on chloride equilibrium concentrations in Na-dominated bentonite. Adding the high concentration chloride equilibrium results from Hed12 and Hsi15 to this data (in terms of \(c^\mathrm{int}/c^\mathrm{ext}\)), gives the following picture6 (the 3WL system corresponds to pure montmorillonite of density ~1300 kg/m3, and the 2WL system corresponds to ~1600 kg/m3, as also verified experimentally).
The x-axis shows montmorillonite effective dry density, and applied external concentrations for each data series are color coded, but also listed in the legend. Note that this plot contains mainly all available information for drawing conclusions regarding anion exclusion in interlayers.7 To me, the conclusions that can be drawn are to a large extent opposite to those that have been drawn:
The amount chloride in the simulated 3WL system corresponds roughly to measured values. Consequently, MD simulations do not support models that completely exclude anions from interlayers.
The 3WL results instead suggest that interlayers contain the main contribution of chloride. Interlayers must consequently be handled no matter how many additional pore structures a model contains.
For systems corresponding to 2WL interlayers, there is a choice: Either,
assume that the discrepancy between simulations and measurements indicates the existence of an additional pore structure, where the majority of chloride resides, or
assume that presently available MD simulations of 2WL systems overestimate “anion” exclusion.8
Tournassat et al. (2016) (Tou16, in the following) present more MD simulations of interlayer pores in contact with an external compartment, with a fixed amount of excess ions, at three different interlayer distances: 2WL (external concentration ~0.5 M), 3WL (~0.4 M), and 5WL (~0.3 M).
In the 2WL simulations, no anions enter the interlayers. Tou16 do not reflect on the possibility that 2WL simulations may overestimate exclusion, as suggested by Hsi159, but instead use this result to argue that anions are basically completely excluded from 2WL interlayers. They even imply that the result of Rot07 is more adequate than that of Hsi15
In the case of the 2WL hydrate, no Cl– ion entered the interlayer space during the course of the simulation, in agreement with the modeling results of Rotenberg et al. (2007b), but in disagreement with those of Hsiao and Hedström (2015).
But, as discussed, there is no real “disagreement” between the
results of Hsi15 and
Rot07. To refute
the conclusions of Hsi15, Tou16 are
required to demonstrate well converged results, and analyze what is
supposedly wrong with the simulations of
Hsi15. It is,
furthermore, glaringly obvious that most of the anion equilibrium
results in Tou16
are not converged.
Regarding convergence, the only “analysis” provided is the following
passage
The simulations were carried out at the same temperature (350 K) as the simulations of Hsiao and Hedström (2015) and with similar simulation times (50 ns vs. 100-200 ns) and volumes (27 × 104 Å3vs. 15 × 104 Å3), thus ensuring roughly equally reliable output statistics. The fact that Cl– ions did not enter the interlayer space cannot, therefore, be attributed to a lack of convergence in the present simulation, as Hsiao and Hedström have postulated to explain the difference between their results and those of Rotenberg et al. (2007b).
I mean that this is not a suitable procedure in a scientific
publication — the authors should of course demonstrate convergence of
the simulations actually performed! (Especially after
Hsi15 have provided
methods for such an analysis.10)
Anyhow, Tou16 completely miss that Hsi15 demonstrate convergence in simulations with external concentration 1.67 M; for the system relevant here (0.55 M), Hsi15 explicitly write that the same level of convergence requires a 10-fold increase of the simulation time (because the interlayer concentration decreases approximately by a factor of 10, as predicted by — Donnan theory). Thus, the simulation time of Tou16 (53 ns) should be compared with 2000 ns, i.e. it is only a few percent of the time required for proper convergence.
Further confirmation that the simulations in
Tou16 are not
converged is given by the data for the systems where chloride
has entered the interlayers. The ion concentration profiles for
the 3WL simulation look like this
The extension of the interlayers is indicated by the dotted lines. Each interlayer was given slightly different (average) surface charge density, which is denoted in the figure. One of the conspicuous features of this plot is the huge difference in chloride content between different interlayers: the concentration in the mid-pore (0.035 M) is more than three times that in left pore (0.010 M). This clearly demonstrates that the simulation is not converged (cf. the converged chloride result of Hsi15). Note further that the larger amount of chloride is located in the interlayer with the highest surface charge, and the least amount is located in the interlayer with the smallest surface charge.11 I think it is a bit embarrassing for Clays and Clay Minerals to have used this plot for the cover page.
As the simulation times (53 ns vs. 40 ns), as well as the external concentrations (~0.5 M vs. ~0.4 M), are similar in the 2WL and and 3WL simulations, it follows from the fact that the 3WL system is not converged, that neither is the 2WL system. In fact, the 2WL system is much less converged, given the considerably lower expected interlayer concentration. This conclusion is fully in line with the above consideration of convergence times in Hsi15.
For chloride in the 3WL (and 5WL) system, Tou16 conclude that “reasonable quantitative agreement was found” between MD and traditional theory, without the slightest mentioning of what that implies.12 I find this even more troublesome than the lack of convergence. If the authors mean that MD simulations reveal the true nature of anion equilibrium (as they do when discussing 2WL), they here pull the rug out from under the entire mainstream bentonite view! With the 3WL system containing a main contribution, interlayers can of course not be modeled as anion-free, as we discussed above. Yet, not a word is said about this in Tou16.
In this blog post I have tried to show that available MD simulations do not, in any reasonable sense, support the assumption that anions are completely excluded from interlayers. Frankly, I see this way of referencing MD studies mainly as an “afterthought”, in attempts to justify the misuse of the exclusion-volume concept. In this light, I am not surprised that Hed12 and Hsi15 have not gained reasonable attention, while Tou16 nowadays can be found referenced to support claims that anions do not have access to “interlayers”.13
Footnotes
[1] I should definitely discuss the “Stern layer” in a future blog post. Update (250113): Stern layers are discussed here.
[2] The view of bentonite (“clay”) in Rotenberg et al. (2007) is strongly rooted in a “stack” concept. What I refer to as an “external compartment” in their simulation, they actually conceive of as a part of the bentonite structure, calling it a “micropore”.
[3] That
Rotenberg et
al. (2007) expresses this view of anion exclusion puzzles me
somewhat, since several of the same authors published a study just a
few years later where Donnan theory was explored in similar systems:
Jardat et al. (2009).
[4] Since the number of chloride ions found in the
interlayer is not correlated with how layer charge is distributed,
we can conclude that the latter parameter is not important for the
process.
[5] The small difference in the two
simulations with 4 chloride ions is thus a coincidence.
[6] I am in the process of
assessing the experimental data, and hope to be able to better
sort out which of these data series are more relevant. So far I have
only looked at — and discarded —
the
study by Muurinen et al. (1988). This study is therefore removed
from the plot.
[7] There are of course severalotherresults that indirectly demonstrate the presence of anions in interlayers. Anyway, I think that the bentonite research community, by now, should have managed to produce better concentration data than this (both simulated and measured).
[8] As the cation (sodium) may give a major contribution to the hydration energy barrier (this is not resolved in Hsiao and Hedström (2015)), it may be inappropriate to refer to this part as “anion” exclusion (remember that it is salt that is excluded from bentonite). It may be noted that sodium actually appear to have a hydration barrier in e.g. the Na/Cs exchange process, which has been explored both experimentally and in MD simulations.
[9] Tournassat et al. (2016) even refer to Hsiao and Hedström (2015) as presenting a “hypothesis” that “differences in solvation energy play an important role in inhibiting the entry of Cl– in the interlayer space”, rather than addressing their expressed concern that the hydration energy cost may be overestimated.
[11] As the interlayers have different surface charge, they are not expected to have identical chloride content. But the chloride content should reasonably decrease with increasing surface charge, and the difference between interlayers should be relatively small.
[12] Here we have to disregard that the “agreement” is not quantitative. It is not even qualitative: the highest chloride content was recorded in the interlayer pore with highest charge, in both the 3WL and the 5WL system.
where \(\phi\) is the porosity of the sample, \(D_c\) is the macroscopic
pore diffusivity of the presumed interlayer domain, and \(\Xi\) is the
ion equilibrium coefficient. \(\Xi\) quantifies the ratio between
internal and external concentrations of the ion under consideration,
when the two compartments are in equilibrium.
where \(\epsilon_\mathrm{eff}\) is the porosity of a presumed bulk water
domain where anions are assumed to reside exclusively, and \(D_p\) is
the corresponding pore diffusivity of this bulk water domain.
We have
discussed earlier
how the homogeneous mixture and the effective porosity models can be
equally well fitted to a specific set of anion through-diffusion
data. The parameter “translation” is simply
\(\phi\cdot \Xi \leftrightarrow \epsilon_\mathrm{eff}\) and
\(D_c \leftrightarrow D_p\). It may appear from this equivalency that
diffusion data alone cannot be used to discriminate between the two
models.
But note that the interpretation of how \(D_e\) varies with background
concentration is very different in the two models.
In the homogeneous mixture model, \(D_c\) is not expected to vary with background concentration to any greater extent, because the diffusing domain remains essentially the same. \(D_e\) varies in this model primarily because \(\Xi\) varies with background concentration, as a consequence of an altered Donnan potential.
In the effective porosity model, \(D_p\) is expected to vary, because the volume of the bulk water domain, and hence the entire domain configuration (the “microstructure”), is postulated to vary with background concentration. \(D_e\) thus varies in this model both because \(D_p\) and \(\epsilon_\mathrm{eff}\) varies.
A simple way of taking into account a varying domain configuration (as in the effective porosity model) is to assume that \(D_p\) is proportional to \(\epsilon_\mathrm{eff}\) raised to some power \(n – 1\), where \(n > 1\). Eq. 2 can then be written
where \(D_0\) is the tracer diffusivity in pure bulk water. Eq. 3 is in the bentonite literature often referred to as “Archie’s law”, in analogy with a similar evaluation in more conventional porous systems. Note that with \(D_0\) appearing in eq. 3, this expression has the correct asymptotic behavior: in the limit of unit porosity, the effective diffusivity reduces to that of a pure bulk water domain.
Eq. 3 shows that \(D_e\) in the effective porosity model is expected to depend non-linearly on background concentration for constant sample density. In contrast, since \(D_c\) is not expected to vary significantly with background concentration, we expect a linear dependence of \(D_e\) in the homogeneous mixture model. Keeping in mind the parameter “translation” \(\phi\cdot\Xi \leftrightarrow \epsilon_\mathrm{eff}\), the prediction of the homogeneous mixture model (eq. 1) can be expressed1
We have thus managed to establish a testable difference between the effective porosity and the homogeneous mixture model (eqs. 3 and 4). This is is great! Making this comparison gives us a chance to increase our process understanding.
Comparison with experiment
Van Loon et al. (2007)
It turns out that the chloride diffusion measurements performed by Van Loon et al. (2007) are accurate enough to resolve whether \(D_e\) depends on “\(\epsilon_\mathrm{eff}\)” according to eqs. 3 or 4. As will be seen below, this data shows that \(D_e\) varies in accordance with the homogeneous mixture model (eq. 4). But, since Van Loon et al. (2007) themselves conclude that \(D_e\) obeys Archie’s law, and hence complies with the effective porosity model, it may be appropriate to begin with some background information.
Van Loon et al. (2007) report three different series of diffusion tests, performed on bentonite samples of density 1300, 1600, and 1900 kg/m3, respectively. For each density, tests were performed at five different NaCl background concentrations: 0.01 M, 0.05 M, 0.1 M, 0.4 M, and 1.0 M. The tests were evaluated by fitting the effective porosity model, giving the effective diffusion coefficient \(D_e\) and corresponding “effective porosity” \(\epsilon_\mathrm{eff}\) (it is worth repeating that the latter parameter equally well can be interpreted in terms of an ion equilibrium coefficient).
Van Loon et al. (2007) conclude that their data complies with eq. 3, with \(n = 1.9\), and provide a figure very similar to this one
Here are compared evaluated values of effective diffusivity and “effective porosity” in various tests. The test series conducted by Van Loon et al. (2007) themselves are labeled with the corresponding sample density, and the literature data is from García-Gutiérrez et al. (2006)2 (“Garcia 2006”) and the PhD thesis of A. Muurinen (“Muurinen 1994”). Also plotted is Archie’s law with \(n\) =1.9. The resemblance between data and model may seem convincing, but let’s take a further look.
Rather than lumping together a whole bunch of data sets, let’s focus on the three test series from Van Loon et al. (2007) themselves, as these have been conducted with constant density, while only varying background concentration. This data is thus ideal for the comparison we are interested in (we’ll get back to commenting on the other studies).
It may also be noted that the published plot contains more data points (for these specific test series) than are reported in the rest of the article. Let’s therefore instead plot only the tabulated data.3 The result looks like this
Here we have also added the predictions from the homogeneous mixture model (eq. 4), where \(D_c\) has been fitted to each series of constant density.
The impression of this plot is quite different from the previous one: it should be clear that the data of Van Loon et al. (2007) agrees fairly well with the homogeneous mixture model, rather than obeying Archie’s law. Consequently, in contrast to what is stated in it, this study refutes the effective porosity model.
The way the data is plotted in the article is reminiscent of Simpson’s paradox: mixing different types of dependencies of \(D_e\) gives the illusion of a model dependence that really isn’t there. Reasonably, this incorrect inference is reinforced by using a log-log diagram (I have warned about log-log plots earlier). With linear axes, the plots give the following impression
This and the previous figure show that \(D_e\) depends approximately linearly on “\(\epsilon_\mathrm{eff}\)”, with a slope dependent on sample density. With this insight, we may go back and comment on the other data points in the original diagram.
García-Gutiérrez et al. (2006) and Muurinen et al. (1988)
The tests by García-Gutiérrez et al. (2006) don’t vary the background concentration (it is not fully clear what the background concentration even is4), and each data point corresponds to a different density. This data therefore does not provide a test for discriminating between the models here discussed.
I have had no access to Muurinen (1994), but by examining the data, it is clear that it originates from Muurinen et al. (1988), which was assessed in detail in a previous blog post. This study provides two estimations of “\(\epsilon_\mathrm{eff}\)”, based on either breakthrough time or on the actual measurement of the final state concentration profile. In the above figure is plotted the average of these two estimations.5
One of the test series in Muurinen et al. (1988) considers variation of density while keeping background concentration fixed, and does not provide a test for the models here discussed. The data for the other two test series is re-plotted here, with linear axis scales, and with both estimations for “\(\epsilon_\mathrm{eff}\)”, rather than the average6
As discussed in the assessment of this study, I judge this data to be too uncertain to provide any qualitative support for hypothesis testing. I think this plot confirms this judgment.
Glaus et al. (2010)
The measurements by Van Loon et al. (2007) are enough to convince me that the dependence of \(D_e\) for chloride on background concentration is furtherevidence for that a homogeneous view of compacted bentonite is principally correct. However, after the publication of this study, the same authors (partly) published more data on chloride equilibrium, in pure Na-montmorillonite and “Na-illite”,7 in Glaus et al. (2010).
This data certainly shows a non-linear relation between \(D_e\) and “\(\epsilon_\mathrm{eff}\)” for Na-montmorillonite, and Glaus et al. (2010) continue with an interpretation using “Archie’s law”. Here I write “Archie’s law” with quotation marks, because they managed to fit the expression to data only by also varying the prefactor. The expression called “Archie’s law” in Glaus et al. (2010) is
where \(A\) is now a fitting parameter. With \(A\) deviating from \(D_0\), this expression no longer has the correct asymptotic behavior as expected when interpreting \(\epsilon_\mathrm{eff}\) as quantifying a bulk water domain (see eq. 3). Nevertheless, Glaus et al. (2010) fit this expression to their measurements, and the results look like this (with linear axes)
Here is also plotted the prediction of the homogeneous mixture model
(eq. 4). For the montmorillonite data, the dependence is
clearly non-linear, while for the “Na-illite” I would say that the
jury is still out.
Although the data for montmorillonite in
Glaus et al. (2010)
is
non-linear, there are several strong arguments for why this is not an
indication that the effective porosity model is correct:
Remember that this result is not a confirmation of the measurements in Van Loon et al. (2007). As demonstrated above, those measurements complies with the homogeneous mixture model. But even if accepting the conclusion made in that publication (that Archie’s law is valid), the Glaus et al. (2010) results do not obey Archie’s law (but “Archie’s law”).
The four data points correspond to background concentrations of 0.1 M, 0.5 M, 1.0 M, and 2.0 M. If “\(\epsilon_\mathrm{eff}\)” represented the volume of a bulk water phase, it is expected that this value should level off, e.g. as the Debye screening length becomes small (Van Loon et al. (2007) argue for this). Here “\(\epsilon_\mathrm{eff}\)” is seen to grow significantly, also in the transition between 1.0 M and 2.0 M background concentration.
These are Na-montmorillonite samples of dry density 1.9 g/cm3. With an “effective porosity” of 0.067 (the 2.0 M value), we have to accept more than 20% “free water” in these very dense systems! This is not even accepted by otherproponents of bulk water in compacted bentonite.
Furthermore, these tests were performed with a background of \(\mathrm{NaClO_4}\), in contrast to Van Loon et al. (2007), who used chloride also for the background. The only chloride around is thus at trace level, and I put my bet on that the observed non-linearity stems from sorption of chloride on some system component.
Insight from closed-cell tests
Note that the issue whether or not \(D_e\) varies linearly with
“\(\epsilon_\mathrm{eff}\)” at constant sample density is equivalent
to whether or not \(D_p\) (or \(D_c\)) depends on background
concentration. This is similar to how presumed concentration
dependencies of the pore diffusivity for simple cations
(“apparent”
diffusivities) have been used to argue for multi-porosity in compacted
bentonite. For cations,
a closer look shows that no such dependency is found in the
literature.
For anions, it is a bit frustrating that the literature data is not
accurate or relevant enough to fully settle this issue (the data of
Van Loon et al. (2007)
is, in my opinion, the best available).
However, to discard the conceptual view underlying the effective porosity model, we can simply use results from closed-cell diffusion studies. In Na-montmorillonite equilibrated with deionized water, Kozaki et al. (1998) measured a chloride diffusivity of \(1.8\cdot 10^{-11}\) m2/s at dry density 1.8 g/cm3.8 If the effective porosity hypothesis was true, we’d expect a minimal value for the diffusion coefficient9 in this system, since \(\epsilon_\mathrm{eff}\) approaches zero in the limit of vanishing ionic strength. Instead, this value is comparable to what we can evaluate from e.g. Glaus et al. (2010) at 1.9 cm3/g, and 2.0 M background electrolyte: \(D_e/\epsilon_\mathrm{eff} = 7.2\cdot 10^{-13}/0.067\) m2/s = \(1.1\cdot 10^{-11}\) m2/s.
That chloride diffuses just fine in dense montmorillonite equilibrated with pure water is really the only argument needed to debunk the effective porosity hypothesis.
Footnotes
[1] Note that \(\epsilon_\mathrm{eff}\) is not a parameter in the homogeneous mixture model, so eq. 4 looks a bit odd. But it expresses \(D_e\) if \(\phi\cdot \Xi\) is interpreted as an effective porosity.
[3] This choice is not critical for the conclusions made in this blog post, but it seems appropriate to only include the data points that are fully described and reported in the article.
[4] García-Gutiérrez et al. (2004) (which is the study compiled in García-Gutiérrez et al. (2006)) state that the samples were saturated with deionized water, and that the electric conductivity in the external solution were in the range 1 — 3 mS/cm.
[5] The data point labeled with a “?” seems to have been obtained by making this average on the numbers 0.5 and 0.08, rather than the correctly reported values 0.05 and 0.08 (for the test at nominal density 1.8 g/cm3 and background concentration 1.0 M).
[6] Admittedly, also the data we have plotted from the original tests in Van Loon et al. (2007) represents averages of several estimations of “\(\epsilon_\mathrm{eff}\)”. We will get back to the quality of this data in a future blog post when assessing this study in detail, but it is quite clear that the estimation based on the direct measurement of stable chloride is the more robust (it is independent of transport aspects). Using these values for “\(\epsilon_\mathrm{eff}\)”, the corresponding plot looks like this
[7] To my mind, it is a misnomer to describe something as illite in sodium form. Although “illite” seems to be a bit vaguely defined, it is clear that it is supposed to only contain potassium as counter-ion (and that these ions are non-exchangeable; the basal spacing is \(\sim\)10 Å independent of water conditions). The material used in Glaus et al. (2010) (and severalotherstudies) has a stated cation exchange capacity of 0.22 eq/kg, which in a sense is comparable to the montmorillonite material (a factor 1/4). Shouldn’t it be more appropriate to call this material e.g. “mixed-layer”?
[8] This value is the average from two tests performed at 25 °C. The data from this study is better compiled in Kozaki et al. (2001).
[9] Here we refer of course to the empirically defined diffusion coefficient, which I have named \(D_\mathrm{macr.}\) in earlier posts. This quantity is model independent, but it is clear that it should be be associated with the pore diffusivities in the two models here discussed (i.e. with \(D_c\) in the homogeneous mixture model, and with \(D_p\) in the effective porosity model).
Mu88 performed both chloride and uranium through-diffusion tests on “MX-80” bentonite, as well as sorption tests. Here we focus solely on the chloride diffusion. We also disregard one diffusion test series that does not vary external concentration (it was conducted with an unspecified “artificial groundwater” and varied sample density).
Left are two test series performed with nominal sample densities 1.2 g/cm3 and 1.8 g/cm3, respectively. For each of these densities, chloride through-diffusion tests were performed with external NaCl concentrations of 0.01 M, 0.1 M, and 1.0 M, respectively. The samples were cylindrical with a diameter of 3.0 cm, and a length of 1.5 cm, giving a volume of 10.6 cm3. To refer to a specific test or sample, we use the nomenclature “nominal density/external concentration”, e.g. the test performed at nominal density 1.2 g/cm3 and external solution 0.1 M is referred to as “1.2/0.1”.
Uncertainty of bentonite samples
“MX-80” is not the name of some specific standardized material, but simply a product name.2 It is quite peculiar that that “MX-80” nevertheless is a de facto standard in the research field for clay buffers in radwaste repositories. But, being a de facto standard, several batches of bentonite with this name have been investigated and reported throughout the years. We consequently have some appreciation for its constitution, and the associated variation.
In Mu88, the material used is only mentioned by name, and it is only
mentioned once (in the abstract!). We therefore can’t tell which of
the studies that is more appropriate to refer to. Instead, let’s take
a look at how “MX-80” has been reported generally.
*) These values were derived from summing the exchangeable ions, and are probably overestimations.
Montmorillonite content
Reported montmorillonite content varies in the range 75 — 85%. For the present context, this primarily gives an uncertainty in adopted effective montmorillonite dry density, which, in turn, is important for making relevant comparison between bentonite materials with different montmorillonite content. For the “MX-80” used in Mu88 we here assume a montmorillonite content of 80%. In the table below is listed the corresponding effective montmorillonite densities when varying the montmorillonite content in the range \(x =\) 0.75 — 0.85, for the two nominal dry densities.
Dry density
EMDD (\(x\)=0.75)
EMDD (\(x\)=0.80)
EMDD (\(x\)=0.85)
(g/cm3)
(g/cm3)
(g/cm3)
(g/cm3)
1.2
1.01
1.05
1.09
1.8
1.61
1.66
1.70
The uncertainty in montmorillonite content thus translates to an
uncertainty in effective montmorillonite dry density on the order of
0.1 g/cm3.
Cation population
While reported values of the cation exchange capacity of “MX-80” are relatively constant, of around 0.75 eq/kg,4 the reported fraction of sodium ions is seen to vary, in the range 70 — 85 %. The remaining population is mainly di-valent rare-earth metal ions (calcium and magnesium). This does not only mean that different studies on “MX-80” may give results for quite different types of systems, as the mono- to di-valent ion ratio may vary, but also that samples within the study may represent quite different systems. We examine this uncertainty below, when discussing the external solutions.
Soluble calcium minerals
The uncertainty of how much divalent cations are available is in fact larger than just discussed. “MX-80” is reported to contain a certain amount of soluble calcium minerals, in particular gypsum. These provide additional sources for divalent ions, which certainly will be involved in the chemical equilibration as the samples are water saturated. Reported values of gypsum content in “MX-80” are on the order of 1%. With a molar mass of 0.172 kg/mol, this contributes to the calcium content by \(2\cdot 0.01/0.172\) eq/kg \(\approx 0.12\) eq/kg, or about 16% of the cation exchange capacity.
Sample density
The samples in Mu88 that we focus on have nominal dry density of 1.2
and 1.8 g/cm3. The paper also reports measured porosities on each
individual sample, listed in the below table together with
corresponding values of dry density5
Test
\(\phi\)
\(\rho_d\)
(-)
(g/cm3)
1.2/0.01
0.54
1.27
1.2/0.1
0.52
1.32
1.2/1.0
0.49
1.40
1.8/0.01
0.37
1.73
1.8/0.1
0.31
1.89
1.8/1.0
0.34
1.81
We note a substantial variation in measured density for samples with the same nominal density: for the 1.2 g/cm3 samples, the standard deviation is 0.06 g/cm3, and for the 1.8 g/cm3 samples it is 0.07 g/cm3. Moreover, while the mean value for the 1.8 g/cm3 samples is close to the nominal value (1.81 g/cm3), that for the 1.2 g/cm3 samples is substantially higher (1.33 g/cm3).
It is impossible to know from the information provided in Mu88 if this
uncertainty is intrinsic to the procedure of preparing the samples, or
if it is more related to the procedure of measuring the density at
test termination.6
Uncertainty of external solutions
Mu88 do not describe how the external solutions were prepared. We
assume here, however, that preparing pure NaCl solutions gives no
significant uncertainty.
Further, the paper contains no information on how the samples were water saturated, nor on the external solution volumes. Since samples with an appreciable amount of di-valent cations are contacted with pure sodium solutions, it is unavoidable that an ion exchange process is initiated. As we don’t know any detail of the preparation process, this introduces an uncertainty of the exact aqueous chemistry during the course of a test.
To illustrate this problem, here are the results from calculating the
exchange equilibrium between a sample initially containing 30%
exchangeable charge in form of calcium (70% sodium), and external
NaCl solutions of various concentrations and volumes
In these calculations we assume a sample of density 1.8 g/cm3 with the
same volume as in Mu88 (10.6 cm3), a cation exchange capacity of 0.75
eq/kg, and a Ca/Na selectivity coefficient of 5.
In a main series, we varied the external volume between 50 and 1000 ml
(solid lines). While the solution volume naturally has a significant
influence on the process, it is seen that the initial calcium content
essentially remain for the lowest concentration (0.01 M). In contrast,
for a 1.0 M solution, a significant amount of calcium is exchanged for
all the solution volumes.
The figure also shows a case for sample density 1.2 g/cm3 (dashed line), and a scenario where equilibrium has been obtained twice, with a replacement of the first solution (to a once again pure NaCl solution) (dot-dashed line).
The main lesson from these simulations is that the actual amount of di-valent ions present during a diffusion test depends on many details: the way samples were saturated, volume of external solutions, if and how often solutions were replaced, time, etc. It is therefore impossible to state the exact ion population in any of the tests in Mu88. But, guided by the simulations, it seems very probable that the tests performed at 0.01 M contain a substantial amount of di-valent ions, while those performed at 1.0 M probably resemble more pure sodium systems.
The only information on external solutions in Mu88 is that the
“solution on the low concentration side was changed regularly”
during the course of a test. This implies that the amount of di-valent
cations may not even be constant during the tests.
Uncertainty of diffusion parameters
The diffusion parameters explicitly listed in Mu88 are \(D_e\) and “\(D_a\)”, while it is implicitly understood that they have been obtained by fitting the effective porosity model to outflux data and the measured clay concentration profile in the final state. “\(D_a\)” is thus really the pore diffusivity \(D_p\),7 and relates to \(D_e\) as \(D_e = \epsilon_\mathrm{eff} D_p\), where \(\epsilon_\mathrm{eff}\) is the so-called “effective porosity”. In a previous blog post, we discussed in detail how anion equilibrium concentrations can be extracted from through-diffusion tests, and the results derived there is used extensively in this section.
Rather than fitting the model to the full set of data (i.e. outflux
evolution and final state concentration profile), diffusion parameters
in Mu88 have been extracted in various limits.
Evaluation of \(D_e\) in Mu88
The effective diffusivity was obtained by estimating the steady-state flux, dividing by external concentration difference of the tracer, and multiplying by sample length \begin{equation} D_e = \frac{j^\mathrm{ss}\cdot L}{c^\mathrm{source}}\tag{1} \end{equation}
Here it is assumed that the target reservoir tracer concentration can
be neglected (we assume this
throughout). Eq. 1 is basically eq. 1 in
Mu88
(and
eq. 8 in the earlier blog post), from which we can evaluate the
values of the steady-state flux that was used for the reported values
of \(D_e\) (\(A \approx 7.1\) cm2 denotes sample cross sectional area)
Test
\(D_e\)
\(A\cdot j^\mathrm{ss}/c^\mathrm{source}\)
(\(\mathrm{m^2/s}\))
(ml/day)
1.2/0.01
\(7.7\cdot 10^{-12}\)
0.031
1.2/0.1
\(2.9\cdot 10^{-11}\)
0.118
1.2/1.0
\(1.2\cdot 10^{-10}\)
0.489
1.8/0.01
\(3.3\cdot 10^{-13}\)
0.001
1.8/0.1
\(4.8\cdot 10^{-13}\)
0.002
1.8/1.0
\(4.0\cdot 10^{-12}\)
0.016
The figure below compares the evaluated values of the steady-state
flux with the flux evaluated from the measured target concentration
evolution,8 for samples with nominal dry
density 1.8 g/cm3 (no concentration data was reported for the 1.2
g/cm3 samples)
These plots clearly show that the transition to steady-state is only
resolved properly for the test with highest background concentration
(1.0 M). It follows that the uncertainty of the evaluated steady-state
— and, consequently, of the evaluated \(D_e\) values — increases
dramatically with decreasing background concentration for these
samples.
Evaluation of \(D_p\) in Mu88
Pore diffusivities were obtained in two different ways. One method was to relate the steady-state flux to the clay concentration profile at the end of the test, giving \begin{equation} D_{p,c} = \frac{j^\mathrm{ss}\cdot L}{\phi\cdot\bar{c}(0)} \tag{2} \end{equation}
where \(\bar{c}(0)\) denotes the chloride clay concentration at the interface to the source reservoir. The quantity in eq. 2 is called “\(D_{ac}\)”7 in Mu88, and this equation is essentially the same as eq. 2 in Mu889 (and eq. 10 in the previous blog post). Using the steady-state fluxes, we can back-calculate the values of \(\bar{c}(0)\) used for this evaluation of \(D_{p,c}\)
Test
\(D_{p,c}\)
\(A\cdot j^\mathrm{ss}/c^\mathrm{source}\)
\(\phi\)
\(\bar{c}(0)/c^\mathrm{source}\)
(\(\mathrm{m^2/s}\))
(ml/day)
(-)
(-)
1.2/0.01
\(7.0\cdot 10^{-11}\)
0.031
0.54
0.204
1.2/0.1
\(2.8\cdot 10^{-10}\)
0.118
0.52
0.199
1.2/1.0
\(5.1\cdot 10^{-10}\)
0.489
0.49
0.480
1.8/0.01
\(2.0\cdot 10^{-11}\)
0.001
0.37
0.045
1.8/0.1
\(3.1\cdot 10^{-11}\)
0.002
0.31
0.050
1.8/1.0
\(5.2\cdot 10^{-11}\)
0.016
0.34
0.226
Note that, although we did some calculations to obtain them, the values for \(\bar{c}(0)/c^\mathrm{source}\) in this table are closer to the actual measured raw data (concentrations). We made the calculation above to “de-derive” these values from the reported diffusion coefficients (combining eqs. 1 and 2 shows that \(\bar{c}(0)\) is obtained from the reported parameters as \(\bar{c}(0)/c^\mathrm{source} = D_e/(\phi D_{p,c})\)).
Here are compared the measured concentration profiles for the samples
of nominal density 1.8 g/cm3 and the corresponding slopes used to
evaluate \(D_{p,c}\) (profiles for the 1.2 g/cm3 samples are not
provided in Mu88)
For background concentrations 1.0 M and 0.1 M, the evaluated slope
corresponds quite well to the raw data. For the 0.01 M sample,
however, the match not very satisfactory. I suspect that a detection
limit may have been reached for the analysis of the profile of this
sample. Needless to say, the evaluated value of \(\bar{c}(0)\) is very
uncertain for the 0.01 M sample.
It may also be noted that all measured concentration profiles deviates from linearity near the interface to the source reservoir. This is a general behavior in through-diffusion tests, which I am quite convinced of is related to sample swelling during dismantling, but there are also other suggestedexplanations. Here we neglect this effect and relate diffusion quantities to the linear parts of profiles, but this issue should certainly be treated in a separate discussion. Update (220407): non-linear profiles are discussed here.
\(D_p\) was also evaluated in a different way in Mu88, by measuring what we here will call the breakthrough time, \(t_\mathrm{bt}\) (Mu88 call it “time-lag”). This quantity is fairly abstract, and relates to the asymptotic behavior of the analytical expression for the outflux that apply for constant boundary concentrations (we here assume them to be \(c^\mathrm{source}\) and 0, respectively). This expression is displayed in eq. 7 in the previous blog post.
Multiplying the outflux by the sample cross sectional area \(A\) and integrating, gives the accumulated amount of diffused tracers. In the limit of long times, this quantity is, not surprisingly, linear in \(t\) \begin{equation} A\cdot j^\mathrm{ss} \cdot \left(t – \frac{L^2}{6\cdot D_p} \right ) \end{equation}
\(t_\mathrm{bt}\) is defined as the time for which this asymptotic
expression is zero. Determining \(t_\mathrm{bt}\) from the measured
outflux evolution consequently allows for an estimation of \(D_p\) as
\begin{equation}
D_{p,t} = \frac{L^2}{6t_\mathrm{bt}} \tag{3}
\end{equation}
This quantity is called “\(D_{at}\)” in Mu887
(eq. 3 is eq. 3 in Mu88). With another back
calculation we can extract the values of \(t_\mathrm{bt}\) determined
from the raw data
Test
\(D_{p,t}\)
\(t_\mathrm{bt}\)
(\(\mathrm{m^2/s}\))
(days)
1.2/0.01
\(1.4\cdot 10^{-10}\)
3.1
1.2/0.1
\(2.0\cdot 10^{-10}\)
2.2
1.2/1.0
\(3.2\cdot 10^{-10}\)
1.4
1.8/0.01
\(5.0\cdot 10^{-11}\)
8.7
1.8/0.1
\(5.4\cdot 10^{-11}\)
8.0
1.8/1.0
\(7.7\cdot 10^{-11}\)
5.6
These evaluated breakthrough times are indicated
in the flux plots above for samples of
nominal dry density 1.8 g/cm3. For the 0.1 M and 0.01 M
samples it is obvious that this value is very uncertain — without a
certain steady-state flux it is impossible to achieve a certain
breakthrough time. The breakthrough time for the 1.8/1.0 test, on
the other hand, simply appears to be incorrectly evaluated: in terms
of outflux vs. time, the breakthrough time should be the time where
the flux has reached 62% of the steady-state
value.10
As no raw data is reported for the 1.2 g/cm3 tests, the quality of the
evaluated breakthrough times cannot be checked for them. It may be
noted, however, that the evaluated breakthrough times are
significantly shorter in this case as compared with the 1.8 g/cm3
tests. Consequently, while the sampling frequency is high enough to
properly resolve the transient stage of the outflux evolution for the
1.8g/cm3 tests, it must be substantially higher in order to resolve
this stage in the 1.2g/cm3 tests (I guess a rule of thumb is that
sampling frequency must be at least higher than \(1/t_{bt}\)).
In a well conducted study these estimates should be similar; \(D_{p,c}\) and \(D_{p,t}\) are, after all, estimations of the same quantity: the pore diffusivity \(D_p\).7 But here we note a discrepancy of approximately a factor 2 between several values of \(\bar{c}(0)\).
It is difficult to judge generally which of the estimations are more
accurate, but we have seen that for the 1.8/0.1 and 1.8/0.01 tests,
the flux data is not very well resolved, giving a
corresponding uncertainty on the equilibrium concentration estimated
from the breakthrough time. On the other hand, also
the concentration profile is poorly
resolved in the case of 0.01 M at 1.8 g/cm3.
However, in cases where the value of \(\bar{c}(0)/c^\mathrm{source}\) is substantial (as for the 1.8/1.0 test and, reasonably, for all tests at 1.2 g/cm3), we expect the estimation directly from the concentration profile to be accurate and robust (as for the 1.8 g/cm3 test at high NaCl concentration). For the 1.2 g/cm3 samples we cannot say much more than this, since Mu88 don’t provide the concentration raw data. For the 1.8/1.0 test, however, we can continue the analysis by fitting the model to all available data.
Re-evaluation by fitting to the full data set
Note that all evaluations in Mu88 are based on making an initial estimation of the steady-state flux, giving \(D_e\) (eq. 1). This value of \(D_e\) (or \(j^{ss}\)) is thereafter fixed in the subsequent estimation of \(D_{p,c}\) (eq. 2). Likewise, an estimation of the steady-state flux is required for estimating the breakthrough time. Here is an animation showing the variation of the model when transitioning from the value of the pore diffusivity estimated from breakthrough time (\(7.7\cdot 10^{-11}\) m2/s), to the value estimated from concentration profile (\(5.2\cdot 10^{-11}\) m2/s) for the 1.8/1.0 test, keeping the steady-state flux fixed at the initial estimation
Note that the axes for the flux is on top (time) and to the right (accumulation rate). This animation confirms that the diffusivity evaluated from breakthrough time in Mu88 gives a way too fast process: the slope of the steady-state concentration profile is too small, and the outflux evolution has a too short transient stage. On the other hand, using the diffusivity estimated from the concentration profiles still doesn’t give a flux that fit very well. The problem is that this fitting is performed with a fixed value of the steady-state flux. By instead keeping the slope fixed at the experimental values, while varying diffusivity (and thus steady state flux), we get the following variation
This animation shows that the model can be fitted well to all data (at least for the 1.8/1.0 test). The problem with the evaluation in Mu88 is that it assumes the steady-state to be fully reached at the later stages of the test. As the above fitting procedure shows, this is only barely true. The experiments could thus have been designed better by conducting them longer, in order to better sample the steady-state phase (and the steady-state flux should have been fitted to the entire data set). Nevertheless, for this sample, the steady-state flux obtained by allowing for this parameter to vary is only slightly different from that used in Mu88 (17.5 rather than 16.3 \(\mathrm{\mu}\)l/day, corresponding to a change of \(D_p\) from \(5.2\cdot10^{-11}\) to \(5.6\cdot10^{-11}\) m2/s). Moreover, this consideration should not be a problem for the 1.2 g/cm3 tests, if they were conducted for as long time as the 1.8 g/cm3 tests, because steady-state is reached much faster (in those tests, sampling frequency may instead be a problem, as discussed above).
As we were able to fit the full model to all data, we conclude that the value of \(\bar{c}(0)/c^\mathrm{source}\) obtained from \(D_{p,c}\) is probably the more robust estimation11, and that there appears to be a problem with how the breakthrough times have been determined. For the 1.8 g/cm3 samples we have demonstrated that this is the case, for the 1.2 g/cm3 we can only make an educated guess that this is the case.
Summary and verdict
We have seen that the results on chloride diffusion in Mu88 suffer from uncertainty from several sources:
The “MX-80” material is not that well defined
Densities vary substantially for samples at the same nominal density
Without knowledge of e.g water saturation procedures and solution volumes, it is impossible to estimate the proper ion population during the course of a test
It is, however, highly likely that tests performed at low NaCl concentrations contain substantial amounts of di-valent ions, while those at high NaCl concentration are closer to being pure sodium systems.
The reported diffusivities give a corresponding uncertainty in the chloride equilibrium concentrations of about a factor of 2. While some tests essentially have a too high noise level to give certain estimations, the problem for the others seems to stem from the estimation of breakthrough times.
Here is an attempt to encapsulate the above information in an
updated plot for the chloride equilibrium data in Mu88
The colored squares represent “confidence areas” based on the variation within each nominal density (horizontally), and on the variation of \(\bar{c}(0)/c^\mathrm{source}\) from the two reported values on pore diffusivity7 (vertically). The limits of these rectangles are simply the 95% confidence interval, based on these variations, and assuming a normal distribution.
Data points put within parentheses are estimations judged to be
improper (based on either re-evaluation of the raw data, or informed
guesses).
From the present analysis my decision is to not use the data
from Mu88 to e.g. validate models for anion exclusion. Although there
seems to be nothing fundamentally wrong with how these test were
conducted, they suffer from so many uncertainties of various sources
that I judge the data to not contribute to quantitative process
understanding.
[2] MX-80 is not only a brand name, but also
a band
name.
[3] This report is “Bentonite Mineralogy” by L. Carlson (Posiva WR 2004-02), but it appears to not be included in the INIS database. It can, however, be found with some elementary web searching.
[4] It’s interesting to note that the cation exchange capacity of
“MX-80” remains more or less constant, while the montmorillonite
content has some variation. This implies that the montmorillonite
layer charge varies (and is negatively correlated with montmorillonite
content). Could it be that the manufacturer has a specified cation
exchange capacity as requirement for this product?
[5] To convert porosity to
dry density, I used \(\rho_d = \rho_s\cdot(1-\phi)\), with solid grain
density \(\rho_s = 2.75\) g/cm3.
[6] A speculation is that the uncertainty stems from the measurement procedure, as this was done on smaller sections of the full samples. It is not specified in Mu88 what the reported porosity represent, but it is reasonable to assume that it is the average of all sections of a sample.
[8] These values were not tabulated, but I have read
them off from the graphs in Mu88.
[9] Mu88 use the
concentration based on the total volume in their expression, while
\(\bar{c}\) is
defined in terms of water volume (water mass,
strictly). Eq.2 therefore contains the physical
porosity. In their concentration profile plots, however, Mu88 use
\(\bar{c}\) as variable (called \(c_{pw}\) — the “concentration in the pore
water”)
[10] Plugging the breakthrough time \(L^2/6D_p\) into the expression for the flux gives
I find it amusing that this value is close to the reciprocal golden ratio (0.618033…). Finding the breakthrough time from a flux vs. time plot thus corresponds (approximately) to splitting the y-axis according to the golden ratio.
[11] Note that the actual evaluated values of $D_{p,c}$ in Mu88 still may be uncertain, because they also depend on the values of the steady-state flux, which we have seen were not optimally evaluated.
On the surface, “Ionendiffusion in Hochverdichtetem Bentonit”1 by G. Kahr, R. Hasenpatt, and M. Müller-Vonmoos, published by NAGRA in March 1985, looks like an ordinary mundane 37-page technical report. But it contains experimental results that could have completely changed the history of model development for compacted clay.
Test principles
The tests were conducted in a quite original manner. By compacting
granules or powder, the investigators obtained samples that
schematically look like this
The bentonite material — which was either Na-dominated “MX-80”, or Ca-dominated “Montigel” — was conditioned to a specific water-to-solid mass ratio \(w\). At one of the faces, the bentonite was mixed with a salt (in solid form) to form a thin source for diffusing ions. This is essentially the full test set-up! Diffusion begins as soon as the samples are prepared, and a test was terminated after some prescribed amount of time, depending on diffusing ion and water content. At termination, the samples were sectioned and analyzed. In this way, the investigators obtained final state ion distributions, which in turn were related to the initial states by a model, giving the diffusion coefficients of interest.
Note that the experiments were conducted without exposing samples to a liquid (external) solution; the samples were “unsaturated” to various degree, and the diffusing ions dissolve within the bentonite. The samples were not even confined in a test cell, but “free-standing”, and consequently not under pressure. They were, however, stored in closed vessels during the course of the tests, to avoid changes in water content.
With this test principle a huge set of diffusion tests were
performed, with systematic variation of the following variables:
Bentonite material (“MX-80” or “Montigel”)
Water-to-solid mass ratio (7% — 33%)
Dry density (1.3 g/m3 — 2.1 g/m3 )
Diffusing salt (SrCl2, SrI2, CsCl, CsI, UO2(NO3)2, Th(NO3)4, KCl, KI, KNO3, K2SO4, K2CO3, KF)
Distribution of water in the samples
From e.g. X-ray diffraction (XRD) we know that bentonite water at low water content is distributed in distinct, sub-nm thin films. For simplicity we will refer to all water in the samples as interlayer water, although some of it, reasonably, forms interfaces with air. The relevant point is that the samples contain no bulk water phase, but only interfacial (interlayer) water.
I argueextensively on this blog for that interlayer water is the only relevant water phase also in saturated samples under pressure. In the present case, however, it is easier to prove that this is the case, as the samples are merely pressed bentonite powder at a certain water content; the bentonite water is not pressurized, the samples are not exposed to liquid bulk water, nor are they in equilibrium with liquid bulk water. Since the water in the samples obviously is mobile — as vapor, but most reasonably also in interconnected interlayers — it is a thermodynamic consequence that it distributes as to minimize the chemical potential.
There is a ton of literature on how the montmorillonite basal spacing
varies with water content. Here, we use the neat result from
Holmboe et al. (2012)
that the average interlayer distance varies basically
linearly2 with water content, like this
XRD-studies also show that bentonite water is distributed in rather distinct hydration states, corresponding to 0, 1, 2, or 3 monolayers of water.3 We label these states 0WL, 1WL, 2WL, and 3WL, respectively. In the figure is indicated the approximate basal distances for pure 1WL (12.4 Å), 2WL (15.7 Å), and 3WL (19.0 Å), which correspond roughly to water-to-solid mass ratios of 0.1, 0.2, and 0.3, respectively.
From the above plot, we estimate roughly that the driest samples in
Kahr et al. (1985)
(\(w \sim 0.1\)) are in pure 1WL states, then transitions to a mixture
of 1WL and 2WL states (\(w\sim 0.1 – 0.2\)), to pure 2WL states
(\(w \sim 0.2\)), to a mixture of 2WL and 3WL states
(\(w\sim 0.2 – 0.3\)), and finally to pure 3WL states (\(w\sim 0.3\)).
Results
With the knowledge of how water is distributed in the samples, let’s
take a look at the results of
Kahr et al. (1985).
Mobility of interlayer cations confirmed
The most remarkable results are of qualitative character. It is, for
instance, demonstrated that several cations diffuse far into the
samples. Since the samples only contain interlayer water, this is a
direct proof of ion mobility in the interlayers!
Also, cations are demonstrated to be mobile even when the water
content is as low as 7 or 10 %! As such samples are dominated by 1WL
states, this is consequently evidence for ion mobility in 1WL states.
A more quantitative assessment furthermore shows that the cation diffusivities varies with water content in an almost step-wise manner, corresponding neatly to the transitions between various hydration states. Here is the data for potassium and strontium
This behavior further confirms that the ions diffuse in interlayers,
with an increasing diffusivity as the interlayers widen.
It should also be noted that the evaluated values of the diffusivities
are comparable to — or even larger4 — than
corresponding results from saturated, pressurized tests.
This strongly suggests that interlayer diffusivity dominates also in
the latter types of tests, which also has been
confirmedin more recent years. The
larger implication is that interlayer diffusion is the only relevant
type of diffusion in general in compacted bentonite.
Anions enter interlayers (and are mobile)
The results also clearly demonstrate that anions (iodide) diffuse in systems with water-to-solid mass ratio as low as 7%! With no other water around, this demonstrates that anions diffuse in — and consequently have access to — interlayers. This finding is strongly confirmed by comparing the \(w\)-dependence of diffusivity for anions and cations. Here is plotted the data for iodide and potassium (with the potassium diffusivity indicated on the right y-axis)
The iodide mobility increases as the system transitions from 1WL to 2WL, in a very similar way as for potassium (and strontium). If this is not a proof that the anion diffuse in the same domain as the cation I don’t know what is! Also for iodide the value of the diffusivity is comparable to what is evaluated in water saturated systems under pressure, which implies that interlayer diffusivity dominates generally in compacted bentonite, also for anions.
Dependence of diffusivity on water content and density
A conclusion made in
Kahr et al. (1985),
that I am not sure I fully agree with, is that diffusivity mainly
depends on water content rather than density. As seen in the diagrams
above, the spread in diffusivity is quite substantial for a given
value of \(w\). There is actually some systematic variation here: for
constant \(w\), diffusivity tend to increase with dry density.
Although using unsaturated samples introduces additional variation, the present study provides a convenient procedure to study diffusion in systems with very low water content. A more conventional set-up in this density limit has to deal with enormous pressures (on the order of 100 MPa).
Interlayer chemistry
An additional result is not acknowledged in the report, but is a direct consequence of the observations: the tests demonstrate that interlayers are chemically active. The initially solid salt evidently dissolves before being able to diffuse. Since these samples are not even close to containing a bulk water phase (as discussed above), the dissolution process must occur in an interlayer. More precisely, the salt must dissolve in interface water between the salt mineral and individual montmorillonite layers, as illustrated here
This study seems to have made no impact at all
In the beginning of 1985, the research community devoted to radioactive waste barriers seems to have been on its way to correctly identify diffusion in interlayers as the main transport mechanism, and to recognize how ion diffusion in bentonite is influenced by equilibrium with external solutions.
Already in 1981,
Torstenfelt et al. (1981)
concluded that the
traditional diffusion-sorption model is not valid,
for e.g. diffusion of Sr and Cs, in compacted bentonite. They also
noted, seemingly without realizing the full importance, that these
ions diffused even in unsaturated samples with as low water-to-solid
mass ratio as 10%.
A significant diffusion was observed for Sr in dry clay, although slower than for water saturated clay, Figure 4, while Cs was almost immobile in the dry clay.
A year later also
Eriksen and Jacobsson (1982)
concluded that the traditional diffusion model is not valid. They
furthermore pointed out the subtleties involved when interpreting
through-diffusion experiments, due to ion equilibrium effects
One difficulty in correlating the diffusivities obtained from profile analysis to the diffusivities calculated from steady state transport data is the lack of knowledge of the tracer concentration at the solution-bentonite interface. This concentration is generally higher for sorbing species like positive ions (counterions to the bentonite) and lower for negative ions (coions to the bentonite) as shown schematically in figure 11. The equilibrium concentration of any ion in the bentonite and solution respectively is a function of the ionic charge, the ionic strength of the solution and the overall exchanger composition and thereby not readily calculated
By regarding the clay-gel as a concentrated electrolytic system Marinsky has calculated (30) distribution coefficients for Sr2+ and Cs+ ions in good agreement with experimentally determined Kd-values. The low anionic exchange capacity and hence the low anion concentration in the pore solution caused by Donnan exclusion also explain the low concentrations of anionic tracers within the clay-gel
[…]
For simple cations the ion-exchange process is dominating and there is, as also pointed out by Marinsky (30), no need to suppose that the counterions are immobilized. It ought to be emphasized that for the compacted bentonite used in the diffusion experiments discussed in this report the water content corresponds roughly to 2-4 water molecule layers (31). There is therefore really no “free water” and the measured diffusivity \(\bar{D}\) can be regarded as corresponding approximately to the diffusivity within the adsorbed phase […]
Furthermore, also
Soudek et al. (1984)
had discarded the traditional diffusion-sorption model, identified the
exchangeable cations as giving a dominating contribution to mass
transfer, and used Donnan equilibrium calculations to account for the
suppressed internal chloride concentration.
In light of this state of the research front, the contribution of Kahr et al. (1985) cannot be described as anything but optimal. In contrast to basically all earlier studies, this work provides systematic variation of several variables (most notably, the water-to-solid ratio). As a consequence, the results provide a profound confirmation of the view described by Eriksen and Jacobsson (1984) above, i.e. that interlayer pores essentially govern all physico-chemical behavior in compacted bentonite. A similar description was later given by Bucher and Müller-Vonmoos (1989) (though I don’t agree with all the detailed statements here)
There is no free pore water in highly compacted bentonite. The water in the interlayer space of montmorillonite has properties that are quite different from those of free pore water; this explains the extremely high swelling pressures that are generated. The water molecules in the interlayer space are less mobile than their free counterparts, and their dielectric constant is lower. The water and the exchangeable cations in the interlayer space can be compared to a concentrated salt solution. The sodium content of the interlayer water, at a water content of 25%, corresponds approximately to a 3-n salt solution, or six times the concentration in natural seawater. This more or less ordered water is fundamentally different from that which engineers usually take into account; in the latter case, pore water in a saturated soil is considered as a freely flowing fluid. References to the porosity in highly compacted bentonite are therefore misleading. Highly compacted bentonite is an unfamiliar material to the engineer.
Given this state of the research field in the mid-80s, I find it
remarkable that history took a different turn. It appears as the
results of
Kahr et al. (1985)
made no impact at all (it may be noticed that they themselves analyzed
the results in terms of the traditional diffusion-sorption
model). And rather than that researchers began identifying that
transport in interlayers is the only relevant contribution, the
so-called surface diffusion model gained popularity (it was already promoted by
e.g.
Soudek et al. (1984)
and
Neretnieks and Rasmuson (1983)). Although this
model emphasizes mobility of the exchangeable cations, it is still
centered around the idea that compacted bentonite contains bulk
water.5 Most
modern bentonite models
suffer from similar flaws: they are formulated in terms of bulk water,
while many effects related to interlayers are treated as irrelevant or
optional.
For the case of anion diffusion the historical evolution is maybe even more disheartening. In 1985 the notions of “effective” or “anion-accessible” porosities seem to not have been that widely spread, and here was clear-cut evidence of anions occupying interlayer pores. But just a few years later the idea began to grow that the pore space in compacted bentonite should be divided into regions which are either accessible or inaccessible to anions. As far as I am aware, the first use of the term “effective porosity” in this context was used by Muurinen et al. (1988), who, ironically, seem to have misinterpreted the Donnan equilibrium approach presented by Soudek et al. (1984). To this day, this flawed concept is central in many descriptions of compacted clay.
Footnotes
[1] “Ion
diffusion in highly compacted bentonite”
[2] Incidentally, the slope of this line corresponds to a water “density” of 1.0 g/cm3.
[3] This is the region of swelling often
referred to as
“crystalline”.
[4] I’m not sure the evaluation in Kahr et al. (1985) is fully correct. They use the solution to the diffusion equation for an impulse source (a Gaussian), but, to my mind, the source is rather one of constant concentration (set by the solubility of the salt). Unless I have misunderstood, the mathematical expression to be fitted to data should then be an erfc-function, rather than a Gaussian. Although this modification would change the numerical values of the evaluated diffusion coefficients somewhat, it does not at all influence the qualitative insights provided by the study.
[5] I have discussed the surface diffusion model in some detail in previousblogposts.
Repulsion between surfaces and anions is not really the
point
Many publications dealing with “anion” exclusion in compacted bentonite describe the phenomenon as being primarily due to electrostaticrepulsion of anions from the negativelychargedclaysurfaces. This explanation, which may seem plausible both at a first and a second glance, is actually not that satisfactory. There are two major issues to consider:
Although it is popular to use the word “anion” when referring to the phenomenon, it must be remembered that the anions are accompanied by cations, in order to maintain overall charge neutrality; it really is salt that is excluded from the bentonite. This observation shows that the above “explanation” is incomplete: it can be argued with the same logic that salt should accumulate, because the clay surfaces attract the cations of the external salt.
Salt exclusion occurs generally in Donnan systems, also in those that lack surfaces. Its principal explanation can consequently not involve the presence of surfaces. For a simpler system, e.g. potassium ferrocyanide, the “explanation” above translates to claiming that exclusion is caused by “anions” being electrostatically repelled by the ferrocyanide ions. In this case it may be easier to spot the shortcoming of such a claim, and to consider also the potassium ions (which attract anions), as well as the role played by the cations of the excluded salt.
What, then, is the primary cause for salt exclusion? Let us continue with using potassium ferrocyanide as an example of a simple Donnan system, and then translate our findings to the case of compacted bentonite.
Ferrocyanide
Consider a potassium ferrocyanide solution separated from a potassium
chloride solution by a membrane permeable to all but the ferrocyanide
ions. The ionic configuration near the membrane then looks something
like this
Because potassium ions can pass the membrane, and because they have an entropic driving force to migrate out of the ferrocyanide solution, a (microscopic) region is formed in the external solution next to the membrane, with an excess amount of positive charge. Similarly, a region is formed next to the membrane in the ferrocyanide solution with an excess amount of negative charge. Thus, a region of charge separation exists across the membrane — similar to the depletion zone in a p-n junction — over which the electrostatic potential varies. The electric field (= a varying potential) at the interface acts as to pull back potassium ions towards the ferrocyanide solution. The equilibrium width of the space charge region is set when the diffusive flux is balanced by the flux due to the electric field.
With a qualitative understanding of the electrostatic potential configuration we can now give the most plain answer to what causes “anion” exclusion: it is because of the potential difference across the membrane. Chloride ions behave in the opposite way as compared to potassium, with an entropic driving force to enter the ferrocyanide solution, while being pulled back towards the external solution due to the electric field across the membrane.
Here the mindful reader may perhaps object and point out that the electric field restricting the chloride inflow reasonably originates from the ferrocyanide anions. It thus may seem that “anion” exclusion, after all, is caused by repulsion from other negative charges.
Indeed, electrostatic repulsion of anions requires the “push” of some other negatively charged entity. But note that the potential is constant in the interior of the ferrocyanide solution, and only varies near the membrane. The variation of the potential is caused by separation of charge: chloride is as much “pushed” out of the ferrocyanide solution by the ferrocyanide as it is “pulled” out of it, due to electrostatic attraction, by the excess potassium on the other side. Repulsion between charges of equal sign occurs also in the interior of the ferrocyanide solution (or in any ionic solution), but does not in itself lead to salt exclusion.
Bentonite
The above description can be directly transferred to the case of compacted bentonite. Replacing the potassium ferrocyanide with e.g. K-montmorillonite, salt exclusion occurs mainly because potassium can migrate out of the clay region, while montmorillonite particles cannot. Again, we have charge separation with a resulting varying electrostatic potential across the interface.
Admittedly, the general situation is more complicated in bentonite because of the extension of montmorillonite particles; viewed as “anions”, these are irregularly shaped macromolecules with hundreds or thousands of charge centers.
The ion configuration in a bentonite suspension therefore looks quite different from a corresponding ordinary solution, as the montmorillonite charge obviously is constrained to individual particles. Dilute systems thus have charge separation on the particle scale and show salt exclusion even without charge separation at the interface to the external solution. These types of systems (suspensions) have historically been the subject of moststudies on “anion”exclusion, and are usually treated theoretically using the Gouy-Chapman model.
With increasing density, however, the effect of a varying potential between montmorillonite particles diminishes, while the effect of charge separation at the interface increases. For dense systems (> 1.2 g/cm3, say), we may therefore approximate the internal potential as constant and only consider the variation across the interface to the external solution using Donnan’s “classical” framework.1
Here is an illustration of the validity of this approximation:
The figure shows the difference between the external (green) and the average internal (orange) potentials in a 1:1 system of density 1.3 g/cm3 and with external concentration 0.1 M, calculated using Donnan’s “classical” equation. Also plotted is the electrostatic potential across the interlayer (blue) as calculated using the Poisson-Boltzmann equation,2 in a similar system (interlayer distance 1 nm). It is clear that the variation of the Poisson-Boltzmann potential from the average is small in comparison with the Donnan potential.
Repulsion between chloride and montmorillonite particles of course occurs everywhere in compacted bentonite, whereas the phenomenon mainly responsible for salt exclusion occurs only near the interfaces. Merely stating electrostatic repulsion as the cause for salt exclusion in compacted bentonite does not suffice, just as in the case of ferrocyanide.
To illustrate that the salt exclusion effect depends critically on exchangeable cations being able to diffuse out of the bentonite, consider the following thought experiment.3 Compacted K-montmorillonite is contacted with a NaCl solution. But rather than having a conventional component separating the solution and the clay, we imagine a membrane that does not allow for the passage of neither potassium nor clay, but that allows for the passage of sodium and chloride. Since potassium is not allowed to diffuse out of the bentonite, no charge separation occurs across the membrane. With no space charge region, the electrostatic potential does not vary and NaCl is not excluded! (to the extent that the Donnan approximation is valid)
A charge neutral perspective
The explanation for “anion” exclusion that we have explored rests on
the formation of a potential difference across the interface region
between bentonite and external solution. But remember that it is salt
— in our example KCl — that is excluded from the bentonite (or the
ferrocyanide solution), and that the cation (K) gains energy by being
transferred from the external to the internal solution. The electrical
work for transferring a unit of KCl is thus zero (which makes sense
since KCl is a charge neutral entity). In this light, it may seem
unsatisfactory to offer the potential difference as the sole
explanation for salt exclusion.
I therefore think that the following kinematic way of reasoning is very helpful. Instead of considering the mass transfer of Cl across the membrane in terms of oppositely directed “electric” and “diffusive” parts, we lump them together with equal amounts of K transfer, giving two equal but oppositely directed fluxes of KCl. Reasonably, the KCl flux into the ferrocyanide solution is proportional to the external ion concentrations
\(A\) is a coefficient accounting for the transfer resistance across the interface region. Requiring the sum of these fluxes to be zero gives the following relation
We can therefore interpret KCl exclusion as an effect of potassium in the clay providing a potential for “out-transfer”, as soon as the chance is given, i.e. when chloride enters from the external solution. From this perspective salt exclusion could maybe be said to be a form of cation “rejection”.
Footnotes
[1]
Note also that the Gouy-Chapman model is not valid in the
high density limit, although it is
applied (or
alluded to)
in this limit in
manypublications.
But e.g. Schofield (1947)
states (about the Gouy-Chapman solution):
[T]he equation is applicable to cases in which the distance
between opposing surfaces considerably exceeds the distance
between neighboring point charges on the surfaces; for there
will then be a range of electrolyte concentrations over which
the radius of the ionic atmosphere is less than the former and
greater than the latter.
This criterion is not met in compacted bentonite, where instead the interlayer distance is comparable to the distance between neighboring charge centers on the surfaces. Invalid application of the Gouy-Chapman model also seems to underlie the flawed but widespread “anion-accessible porosity” concept.
[2] This calculation uses the equations presented in Engström and Wennerström (1978), and assumes no excess ions and a surface charge density of 0.111 \(\mathrm{C/m^2}\). For real consistency this calculation should really be performed with the boundary condition of 0.1 M external concentration. However, since the purpose of the graph is just to demonstrate the sizes of the two potential variations, and since I have yet to acquire a reasonable tool for performing Poisson-Boltzmann calculations with non-zero external concentration, I disregard this inconsistency. Moreover, the continuum assumption of the Poisson-Boltzmann description is anyway beginning to lose its validity at these interlayer distances. Update (220831): Solutions to the Poisson-Boltzmann equation with non-zero external concentration are presented here.
[3] Perhaps this could be done as a Molecular Dynamics
simulation?
At the atomic level, montmorillonite is built up of so-called TOT-layers: covalently bonded sheets of aluminum (“O”) and silica (“T”) oxide (including the right amount of impurities/defects). In my mind, such TOT-layers make up the fundamental particles of a bentonite sample. Reasonably, since montmorillonite TOT-layers vary extensively in size, and since a single cubic centimeter of bentonite contains about ten million billions (\(10^{16}\)), they are generally configured in some crazily complicated manner.
Stack descriptions in the literature
But the idea that the single TOT-layer is the fundamental building
block of bentonite is not shared with many of today’s bentonite
researchers. Instead, you find descriptions like e.g. this one, from
Bacle et al. (2016)
Clay mineral particles consist of stacks of parallel
negatively-charged layers separated by interlayer
nanopores. Consequently, compacted smectite contains two major
classes of pores: interlayer nanopores (located inside the
particles) and larger mesopores (located between the particles).
In compacted rocks, montmorillonite (Mt) forms aggregates
(particles) with 5–20 TOT layers (Segad et al., 2010). A typical
radial size of these particles is of the order of 0.01 to 1
\(\mathrm{\mu m}\). The pore space between Mt particles is referred to
as interparticle porosity. Depending on the degree of compaction,
the interparticle porosity contributes 10 to 30% of the total water
accessible pore space in Mt (Holmboe et al., 2012; Kozaki et al.,
2001).
Such statements show that researchers have something more complex in mind than individual TOT-layers when speaking of “particles”: they are some sort of assemblages of TOT-layers. The quotation of Bacle et al. (2016), using both the terms “stacks” and “particles”, even hints at an idea of a hierarchy of fundamental structures. Such a hierarchy is expressed explicitly in e.g. Navarro et al. (2017), who provide a figure with the caption “Schematic particle arrangement in highly compacted Na-bentonite” that looks similar to this one:
Here it is clear that they differ between “aggregates” (which I’m
not sure is the same thing as “particles”), “stacks”, and
individual TOT-layers (which I assume are represented by the
line-shaped objects). In the following, however, we will use the term
“stack” to refer to any kind of suggested fundamental structure
built up from individual TOT-layers.
The one-sentence version of this blog post is:
Stacks make no sense as fundamental building blocks in models of water saturated, compacted bentonite.
The easiest argument against stacks is, in my mind, to simply work out
the geometrical consequences. But before doing that we will examine
some of the references given to support statements about stacks in
compacted systems. Often, no references are given at all, but when
they are, they usually turn out to be largely irrelevant for the
system under study, or even to support an opposite view.
Inadequate referencing
As an example (of many) of inadequate referencing, we
use the statement above from Churakov et al. (2014) as
a starting point. I think this is a “good” statement, in the sense
that it makes rather precise claims about how compacted bentonite is
supposed to be structured, and provides references for some key
statements, which makes it easier to criticize.
Clay is normally not a homogeneous lamellar material. It might be
better described as a disordered structure of stacks of platelets,
sometimes called tactoids — a tactoid typically consists of 5-20
platelets.19-21
Here the terminology is quite different from the previous quotations: TOT-layers are called “platelets”, and “particles” are called “tactoids”. Still, they use the phrase “stacks of platelets”, so I think we can continue with using “stack” as a sort of common term for what is being discussed.1 We may also note that here is used the word “clay”, rather than “montmorillonite” (as does Bacle et al. (2016)), but it is clear from the context of the article that it really is montmorillonite/bentonite that is discussed.
Anyhow, Segad et al. (2010) do not give much direct information on the claim we investigate, but provide three new references. Two2 of these — Banin (1967) and Shalkevich et al. (2007) — are actually studies on montmorillonite suspensions, i.e. as far away as you can get from compacted bentonite in terms of density; the solid mass fraction in these studies is in the range 1 – 4%.
The average distance between individual TOT-layers in this density limit is comparable with, or even larger than, their typical lateral extension (~100 nm). Therefore, much of the behavior of low density montmorillonite depends critically on details of the interaction between layer edges and various other components, and systems in this density limit behave very differently depending on e.g. ionic strength, cation population, preparation protocol, temperature, time, etc. This complex behavior is also connected with the fact that pure Ca-montmorillonite does not form a sol, while the presence of as little as 10 – 20% sodium makes the system sol forming. The behaviors and structures of montmorillonite suspensions, however, say very little about how the TOT-layers are organized in compacted bentonite.
We have thus propagated from a statement in Churakov et al. (2014), and a similar one in Segad et al. (2010), that montmorillonite in general, in “compacted rocks” forms aggregates of 5 – 20 TOT-layers, to studies which essentially concern different types of materials. Moreover, the actual value of “5 – 20 TOT layers” comes from Banin (1967), who writes
Evidence has accumulated showing that when montmorillonite is
adsorbed with Ca, stable tactoids, containing 5 to 20 parallel
plates, are formed (1). When the mineral is adsorbed with Na, the
tactoids are not stable, and the single plates are separated from
each other.
This source consequently claims that the single TOT-layers are the fundamental units, i.e. it provides an argument against any stack concept! (It basically states that pure Ca-montmorillonite does not form a sol.) In the same manner, even though Segad et al. (2010) make the above quoted statement in the beginning of the paper, they only conclude that “tactoids” are formed in pure Ca-montmorillonite.
The swelling and sedimentation behavior of Ca-montmorillonite is a very interesting question, that we do not have all the answers to yet. Still, it is basically irrelevant for making statements about the structure in compacted — sodium dominated3 — bentonite.
Churakov et al. (2014) also give two references for the statement that the “interparticle porosity” in montmorillonite is 10 – 30% of the total porosity: Holmboe et al. (2012) and Kozaki et al. (2001). This is a bizarre way of referencing, as these two studies draw incompatible conclusions, and since Holmboe et al. (2012) — which is the more adequately performed study — state that this type of porosity may be absent:
At dry density \(>1.4 \;\mathrm{g/cm^3}\) , the average interparticle
porosity for the [natural Na-dominated bentonite and purified
Na-montmorillonite] samples used in this study was found to be
\(1.5\pm1.5\%\), i.e. \(\le 3\%\) and significantly lower than
previously reported in the literature.
Holmboe et al. (2012)
address directly the discrepancy with earlier studies, and suggest
that these were not properly analyzed
The apparent discrepancy between the basal spacings reported by Kozaki et al. (1998, 2001) using Kunipia-F washed Na-montmorillonite, and by Muurinen et al. (2004), using a Na-montmorillonite originating from Wyoming Bentonite MX-80, and the corresponding average basal spacings of the [Na-montmorillonite originating from Wyoming bentonite MX-80] samples reported in this study may partly be due to water contents and partly to the fact that only apparent \(\mathrm{d_{001}}\) values using Bragg’s law, without any profile fitting, were reported in their studies.
If
Kozaki et al. (2001)
should be used to support a claim about “interparticle porosity”, it
consequently has to be done in opposition to — not in conjunction
with —
Holmboe et al. (2012).
It would then also be appropriate for authors to provide arguments for
why they discard the conclusions of
Holmboe et al. (2012).4
Stacks in compacted bentonite make no geometrical sense
The literature is full of fancy figures of bentonite structure involving stacks. A typical example is found in Wu et al. (2018), and looks similar to this:
This illustration is part of a figure with the caption “Schematic representation of the different porosities in bentonite and the potential diffusion paths.”5 The regular rectangles in this picture illustrate stacks that each seems to contain five TOT-layers (I assume this throughout). Conveniently, these groups of five layers have the same length within each stack, while the length varies somewhat between stacks. This is a quite common feature in figures like this, but it is also common that all stacks are given the same length.
Another feature this illustration has in common with others is that the particles are ordered: we are always shown edges of the TOT-layers. I guess this is partly because a picture of a bunch of stacks seen from “the top” would be less interesting, but it also emphasizes the problem of representing the third dimension: figures like these are in practice figures of straight lines oriented in 2D, and the viewer is implicitly required to imagine a 3D-version of this two-dimensional representation.
A “realistic” stack picture
But, even as a 2D-representation, these figures are not representative
of what an actual configuration of stacks of TOT-layers looks like.
Individual TOT-layers have a distinct thickness of about 1 nm, but
varies widely in the other two dimensions.
Ploehn and Liu (2006)
analyzed the size distribution of Na-montmorillonite (“Cloisite
Na+”) using atomic force microscopy, and found an average aspect
ratio of 180 (square-root of basal area divided by thickness). A
representative single “TOT-line” drawn to scale is consequently
quite different from what is illustrated in in most stack-pictures,
and look like this (click on the figure to see it in full size)
In this figure, we have added “water layers” on each side of the TOT-layer (light red), with the water-to-solid volume ratio of 16. Neatly stacking five such units shows that the rectangles in the Wu et al. (2018)-figure should be transformed like this
But this is still not representative of what an assemblage of five
randomly picked TOT-layers would look like, because the size
distribution has a substantial variance. According to
Ploehn and Liu (2006), the
aspect ratio follows approximately a log-normal distribution. If we
draw five values from this distribution for the length of five
“TOT-lines”, and form assemblages, we end up with structures that
look like this:7
These are the kind of units that should fill the bentonite illustrations. They are quite irregularly shaped and are certainly not identical (this would be even more pronounced when considering the third dimension, and if the stacks contain more layers).
It is easy to see that it is impossible to construct a dense structure
with these building blocks, if they are allowed a random
orientation. The resulting structure rather looks something like this
Such a structure evidently has very low density, and are reminiscent of the gel structures suggested in e.g. Shalkevich et al. (2007) (see fig. 7 in that paper). This makes some sense, since the idea of stacks of TOT-layers (“tactoids”) originated from studies of low density structures, as discussed above.
Note that the structure in pictures like that in Wu et al. (2018) has a substantial density only because it is constructed with stacks with an unrealistic shape. But even in these types of pictures is the density not very high: with some rudimentary image analysis we conclude that the density in the above picture is only around 800 kg/m3. Also the figure from Navarro et al. (2017) above gives a density below 1000 kg/m3, although there it is explicitly stated that it is a representation of “highly compacted bentonite”.
The only manner in which the “realistic” building blocks can be
made to form a dense structure is to keep them in the same
orientation. The resulting structures then look e.g. like this
where we have color coded each stack, to remind ourselves that these
units are supposed to be fundamental.
Just looking at this structure of a “stack of stacks” should make it clear how flawed the idea is of stacks as fundamental structural units in compacted bentonite (note also how unrepresentative the stack-pictures found in the literature are). One of many questions that immediately arises is e.g. why on earth the tiny gaps between stacks (indicated by arrows) should remain. This brings us to the next argument against stacks as fundamental units for compacted water saturated bentonite:
What is supposed to keep stacks together?
Compacted bentonite of interest e.g. for sealing in radioactive waste repositories exerts swelling pressure of several MPa when in contact with external water. This osmotic pressure is a consequence of the presence of the mobile exchangeable cations in montmorillonite. Each “realistic” unit that we have imagined above is thus required to be at a huge elevated pressure, and the individual TOT-layers have a strong driving force to separate. And, unless a mechanism is provided for why such a separation is impossible, this is of course what we expect to happen! As far as I am aware, such a separation inhibiting mechanism has never been suggested in any publication that promotes the stack concept in compacted bentonite. To get a feel for the absurdity of this issue, let’s take a new look at the figure from Navarro et al. (2017)
Assuming that this system is in equilibrium with an external water
reservoir at zero pressure (i.e. atmospheric absolute pressure), the
pressure in the compartment labeled “intra-aggregate space” is also
close to zero. On the other hand, in the “stacks” located just a few
nm away, the pressure is certainly above 10 MPa in many places! A
structure like this is obviously not in mechanical equilibrium! (To use
the term “obvious” here feels like such an understatement.)
Implications
To sum up what we have discussed so far, the following picture
emerges. The bentonite literature is packed with descriptions of
compacted water saturated bentonite as built up of stacks as
fundamental units. These descriptions are so commonplace that they
often are not supported by references. But when they are, it seems
that the entire notion is based on misconceptions. In particular,
structures identified in low density systems (suspensions, gels) have
been carried over, without reflection, to descriptions of compacted
bentonite. Moreover, all figures illustrating the stack concept are
based on inadequate representations of what an arbitrary assemblage of
TOT-layers arranged in this way actually would look like. With a
“realistic” representation it quickly becomes obvious that it makes
little sense to base a fundamental unit in compacted systems on the
stack concept.
My impression is that this flawed stack concept underlies the entire
contemporary mainstream view of compacted bentonite, as e.g. expressed
by
Wu et al. (2018):
A widely accepted view is that the total porosity of bentonite
consists of \(\epsilon_ {ip}\) and \(\epsilon_ {il}\) (Tachi and
Yotsuji, 2014; Tournassat and Appelo, 2011; Van Loon et al., 2007).
\(\epsilon_ {ip}\) is a porosity related to the space between the
bentonite particles and/or between the other grains of minerals
present in bentonite. It can further be subdivided into
\(\epsilon_ {ddl}\) and \(\epsilon_ {free}\). The diffuse double layer,
which forms in the transition zone from the mineral surface to the
free water space, contains water, cations and a minor amount of
anions. The charge at the negative outer surface of the
montmorillonite is neutralized by an excess of cations. The free
water space contains a charge-balanced aqueous solution of cations
and anions. \(\epsilon_ {il}\) represents the space between TOT-layers
in montmorillonite particles exhibiting negatively charged
surfaces. Due to anion exclusion effect, anions are excluded from
the interlayer space, but water and cations are present.
This view can be summarized as:
The fundamental building blocks are stacks of TOT-layers
(“particles”, “aggregates”, “tactoids”, “grains”…)
Electric double layers are present only on external
surfaces of the stacks.
Far away from external surfaces — in the “inter-particle” or
“inter-aggregate” pores — the diffuse layers merge with a bulk
water solution
Interlayer pores are defined as being internal to the stacks,
and are postulated to be fundamentally different from the external
diffuse layers; they play by a different set of rules.
I don’t understand how authors can get away with promoting this
conceptual view without supplying reasonable arguments for all of its
assumptions8 — and with such a
complex structure, there are a lot of assumptions.
As already discussed, the geometrical implications of the suggested structure do not hold up to scrutiny. Likewise, there are many argumentsagainst the presence of substantial amounts of bulk water in compacted bentonite, including the pressure consideration above. But let’s also take a look at what is stated about “interlayers” and how these are distinguished from electric double layers (I will use quotation marks in the following, and write “interlayers” when specifically referring to pores defined as internal to stacks).
“Interlayers”
“Interlayers” are often postulated to be completely devoid of anions. We discussed this assumption in more depth in a previous blog post, where we discovered that the only references supplied when making this postulate are based on the Poisson-Boltzmann equation. But this is inadequate, since the Poisson-Boltzmann equation does describe diffuse layers, and predicts anions everywhere.
By requiring anion-free “interlayers”, authors actually claim that the physico-chemistry of “interlayers” is somehow qualitatively different from that of “external surfaces”, although these compartments have the exact same constitution (charged TOT-layer surface + ions + water). But an explanation for why this should be the case is never provided, nor is any argument given for why diffuse layer concepts are not supposed to apply to “interlayers”.9 This issue becomes even more absurd given the strong empirical evidence for that anions actually do reside in interlayers.
The treatment of anions is not the only ad hoc description of “interlayers”. It also seems close to mandatory to describe them as having a maximum extension, and as having an extension independently parameterized by sample density. E.g. the influential models for Na-bentonite of Bourg et al. (2006) and Tournassat and Appelo (2011) both rely on the idea that “interlayers” swell out to a certain volume that is smaller than the total pore volume, but that still depends on density.
In e.g. Bourg et al. (2006), the fraction of “interlayer” pores remains essentially constant at ~78%, as density decreases from 1.57 g/cm3 to 1.27 g/cm3, while the “interlayers” transform from having 2 monolayers of water (2WL) to having 3 monolayers (3WL). This is a very strange behavior: “interlayers” are acknowledged as having a swelling potential (2WL expands to 3WL), but do, for some reason, not affect 22% of the pore volume! Although such a behavior strongly deviates from what we expect if “interlayers” are treated with conventional diffuse layer concepts, no mechanism is provided.
Another type of macabre consequence of defining “interlayer” pores as internal to stacks is that a completely homogeneous system is described has having no interlayer pores (because it has no stacks). E.g. Tournassat and Appelo (2011) write (\(n_c\) is the number of TOT-layers in a stack)
[…] the number of stacks in the \(c\)-direction has considerable influence on the interlayer porosity, with interlayer porosity increasing with \(n_c\) and reaching the maximum when \(n_c \approx 25\). The interlayer porosity halves with \(n_c\) when \(n_c\) is smaller than 3, and becomes zero for \(n_c = 1\).10
It is not acceptable that using the term interlayer should require
accepting stacks as fundamental units. But the usage of the term as
being internal to stacks is so widespread in the contemporary
bentonite literature, that I fear it is difficult to even communicate
this issue. Nevertheless, I am certain that e.g.
Norrish (1954) does not
depend on the existence of stacks when using the term like this:
Fig. 7 shows the relationship between interlayer spacing and water
content for Na-montmorillonite. There is good agreement between the
experimental points and the theoretical line, showing that
interlayer swelling accounts for all, or almost all, of physical
swelling.
The stack view obstructs real discovery
A severe consequence of the conceptual view just discussed is that “stacking number” — the (average) number of TOT-layers that stacks are supposed to contain — has been established as fitting parameter in models that are clearly over-parameterized. An example of this is Tournassat and Appelo (2011), who write11
Our predictive model excludes anions from the interlayer space and
relates the interlayer porosity to the ionic strength and the
montmorillonite bulk dry density. This presentation offers a good
fit for measured anion accessible porosities in bentonites over a
wide range of conditions and is also in agreement with microscopic
observations.
But since anions do reside in interlayers,12 it would be better if the model didn’t fit: an over-parameterized or conceptually flawed model that fits data provides very little useful information.
A similar more recent example is Wu et al. (2018). In this work, a model based on the stack concept is successfully fitted both to data on \(\mathrm{ReO_4^-}\) diffusion in “GMZ” bentonite and to data on \(\mathrm{Cl^-}\) diffusion in “KWK” bentonite, by varying “stacking number” (among other parameters). Again, as the model assumes anion-free “interlayer” pores, a better outcome would be if it was not able to fit the data. Moreover, this paper focuses mainly on the ability of the model, while not at all emphasizing the fact that about ten (!) times more \(\mathrm{ReO_4^-}\) was measured in “GMZ” as compared with \(\mathrm{Cl^-}\) in “KWK”, at similar conditions in certain cases. The latter observation is quite puzzling and is, in my opinion, certainly worth deeper investigation (and I am fully convinced that it is not explained by differences in “stacking number”).
[3] Note that “sodium
dominated” in this context means ~20% or more.
[4] It may be noticed that Kozaki et al. (2001) see no X-ray diffraction peaks for low density samples:
The basal spacing of water-saturated
montmorillonite was determined by the XRD method. […] It was found
that a basal spacing of 1.88 nm, corresponding to the three-water
layer hydrate state […] was not observed before the dry density
reached 1.0 Mg/m3.
My interpretation of this observation is that the diffraction peak has
shifted to even lower reflection angles (in agreement with the
observations
of Holmboe
et al. (2012)), not registered by the equipment. The alternative
interpretation must otherwise be that “stacks” suddenly cease to
exist below 1.0 g/cm3. (Yet,
Kozaki et al. (2001)
continues to use a certain d-value in their analysis, also for densities
below 1.0 g/cm3.)
[5] I have discussed “diffusion
paths” in an
earlier blog post.
This illustration certainly fits that discussion.
[6] A water-to-solid volume ratio of 1 corresponds basically to
interlayers of three monolayers of water (3WL).
[7] To construct these units, I made the additional choice of placing each layer randomly in the horizontal direction, with the constraint that all layers should be confined within the range of the longest one in each unit.
[8] By “get away with” I mean “pass peer-review”, and by “don’t understand” I mean “understand”.
[10] A mathematical remark: if the interlayer porosity “halves with \(n_c\)” (what does that mean?) when \(n_c = 2\) (“smaller than 3”), it is impossible to simultaneously have zero interlayer porosity for \(n_c = 1\) (unless the interlayer porosity is zero for any \(n_c\)).
[11] I guess the word “presentation” here really should be “representation”?
[12] Note that one of the authors of this paper also claims in a later paper that anions do populate 3-waterlayer interlayers, in accordance with the Poisson-Boltzmann equation:
The agreement
between PB calculations and MD simulation predictions was somewhat
worse in the case of the \(\mathrm{Cl^-}\) concentration profiles than
in the case of the \(\mathrm{Na^+}\) profiles (Figure 3), perhaps
reflecting the poorer statistics for interlayer Cl concentrations
[…] Nevertheless, reasonable quantitative agreement was found
(Table 2).




















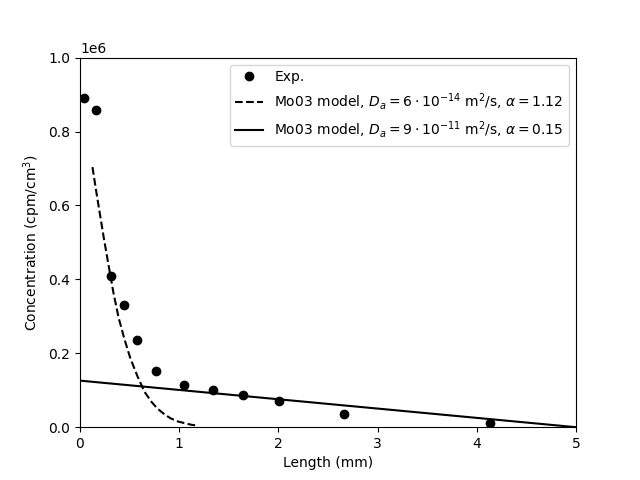





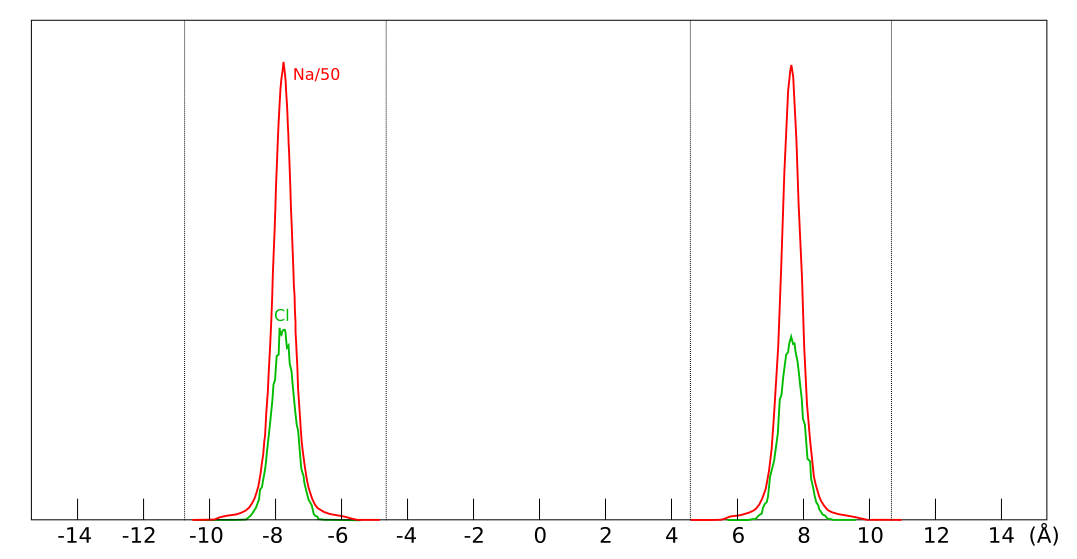









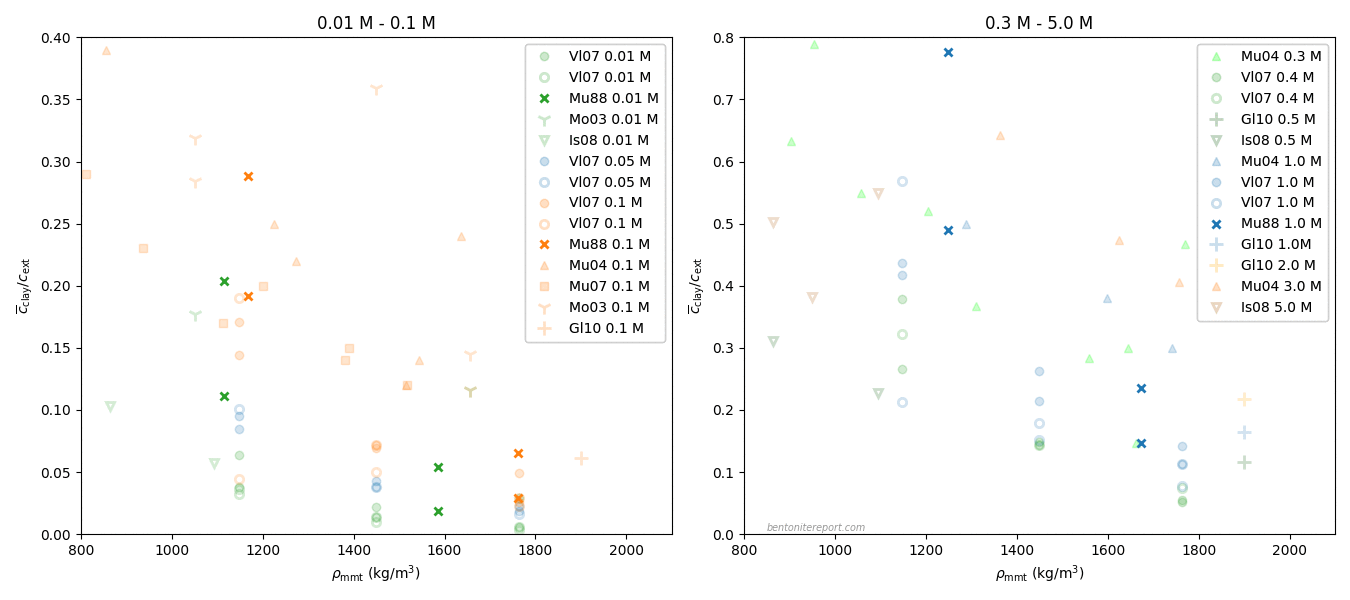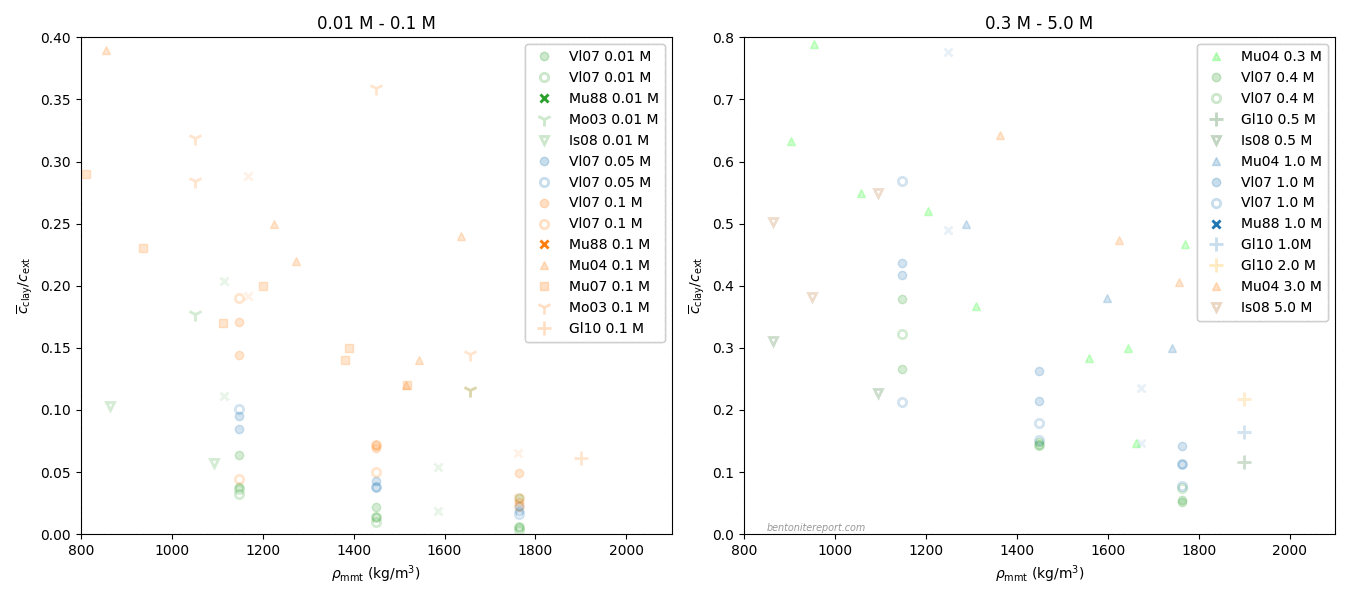

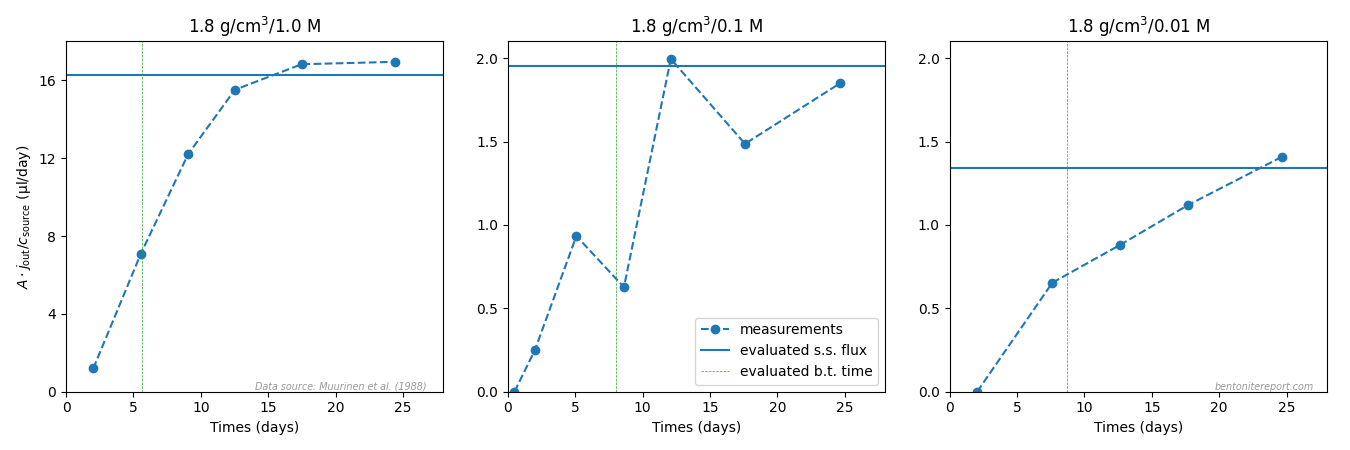







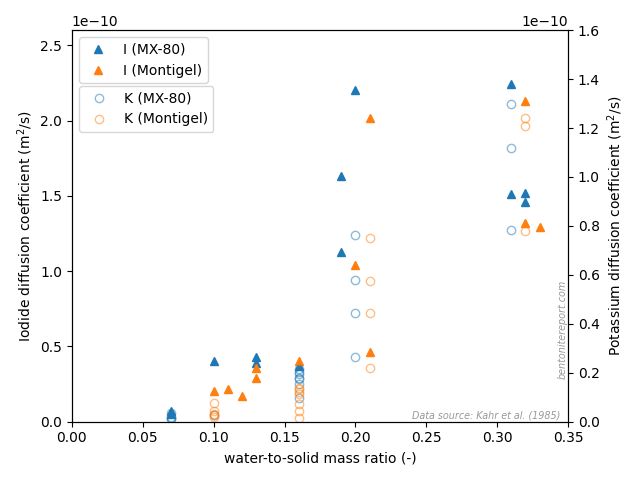


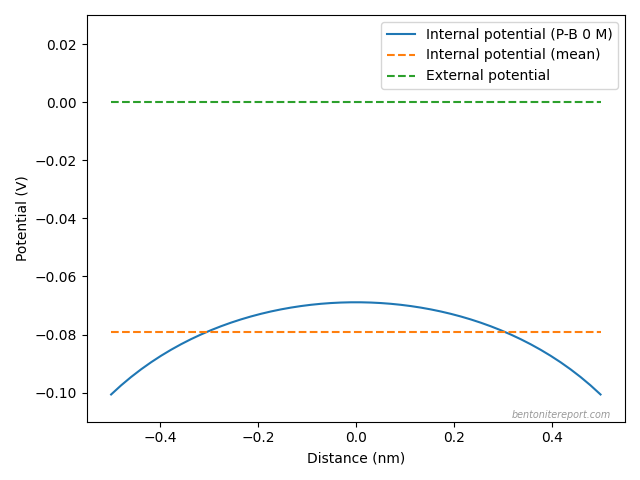

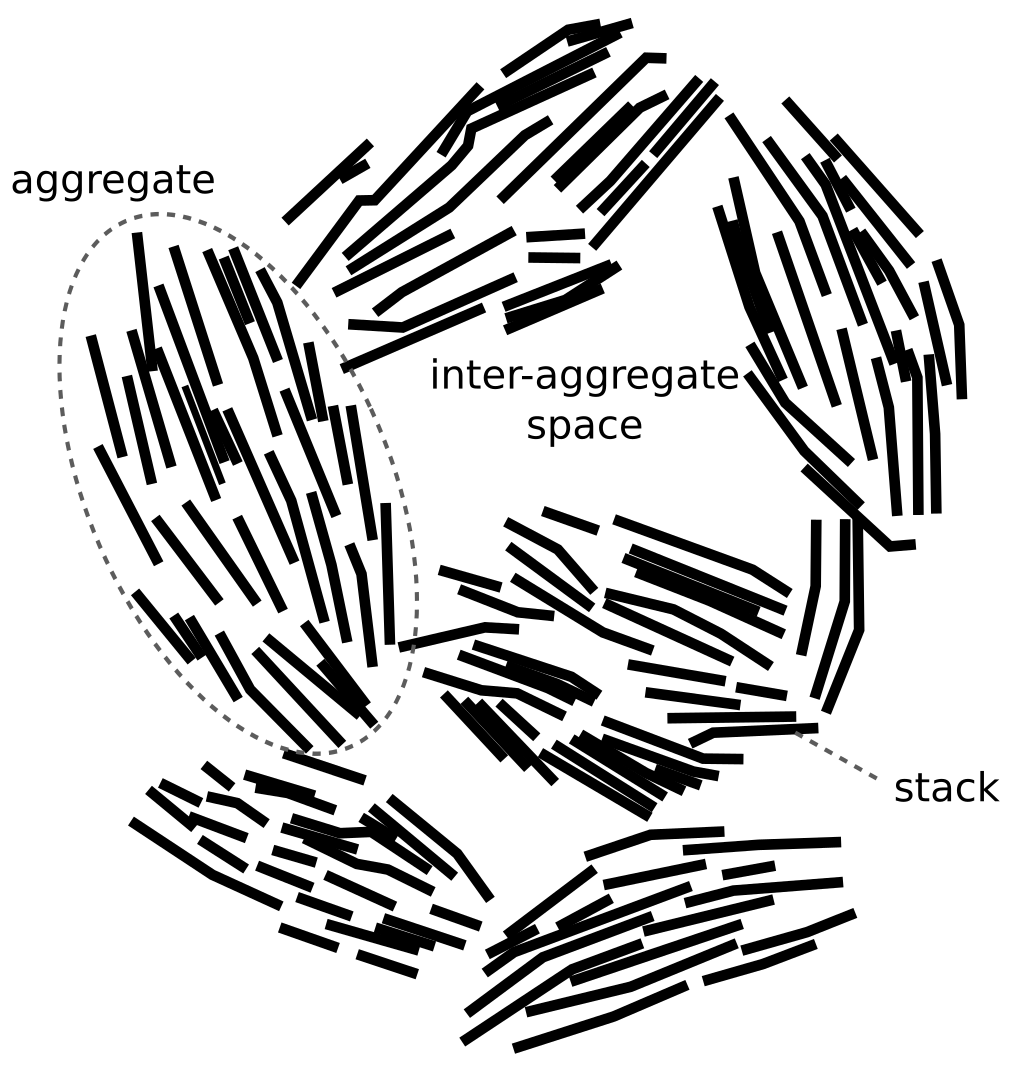







{kind=link}