
Descriptions in bentonite literature
What do authors mean when they say that bentonite has semi-permeable properties? Take for example this statement, from Bradbury and Baeyens (2003)1
[…] highly compacted bentonite can function as an efficient semi-permeable membrane (Horseman et al., 1996). This implies that the re-saturation of compacted bentonite involves predominantly the movement of water molecules and not solute molecules.
Judging from the reference to Horseman et al. (1996) — which we look at below — it is relatively clear that Bradbury and Baeyens (2003) allude to the concept of salt exclusion when speaking of “semi-permeability” (although writing “solute molecules”). But a lowered equilibrium salt concentration does not automatically say that salt is less transferable.
A crucial question is what the salt is supposed to permeate. Note that a semi-permeable component is required for defining both swelling pressure and salt exclusion. In case of bentonite, this component is impermeable to the clay particles, while it is fully permeable to ions and water (in a lab setting, it is typically a metal filter). But Bradbury and Baeyens (2003) seem to mean that in the process of transferring aqueous species between an external reservoir and bentonite, salt is somehow effectively hindered to be transferred. This does not make much sense.
Consider e.g. the process mentioned in the quotation, i.e. to saturate a bentonite sample with a salt solution. With unsaturated bentonite, most bets are off regarding Donnan equilibrium, and how salt is transferred depends on the details of the saturation procedure; we only know that the external and internal salt concentrations should comply with the rules for salt exclusion once the process is finalized.
Imagine, for instance, an unsaturated sample containing bentonite pellets on the cm-scale that very quickly is flushed with the saturating solution, as illustrated in this state-of-the-art, cutting-edge animation

The evolution of the salt concentration in the sample will look something like this

Initially, as the saturating solution flushes the sample, the concentration will be similar to that of the external concentration (\(c_\mathrm{ext}\)). As the sample reaches saturation, it contains more salt than what is dictated by Donnan equilibrium (\(c_\mathrm{eq.}\)), and salt will diffuse out.
In a process like this it should be obvious that the bentonite not in any way is effectively impermeable to the salt. Note also that, although this example is somewhat extreme, the equilibrium salt concentration is probably reached “from above” in most processes where the clay is saturated with a saline solution: too much salt initially enters the sample (when a “microstructure” actually exists) and is later expelled.
Also for mass transfer between an external solution and an already saturated sample does it not make sense to speak of “semi-permeability” in the way here discussed. Consider e.g. a bentonite sample initially in equilibrium with an external 0.3 M NaCl solution, where the solution suddenly is switched to 1.0 M. Salt will then start to diffuse into the sample until a new (Donnan) equilibrium state is reached. Simultaneously (a minute amount of) water is transported out of the clay, in order for the sample to adapt to the new equilibrium pressure.2

There is nothing very “semi-permeabilic” going on here — NaCl is obviously free to pass into the clay. That the equilibrium clay concentration in the final state happens to be lower than in the external concentration is irrelevant for how how difficult it is to transfer the salt.
But it seems that many authors somehow equate “semi-permeability” with salt exclusion, and also mean that this “semi-permeability” is caused by reduced mobility for ions within the clay. E.g. Horseman et al. (1996) write (in a section entitled “Clays as semi-permeable membranes”)
[…] the net negative electrical potential between closely spaced clay particles repel anions attempting to migrate through the narrow aqueous films of a compact clay, a phenomenon known as negative adsorption or Donnan exclusion. In order to maintain electrical neutrality in the external solution, cations will tend to remain
with their counter-ions and their movement through the clay will also be restricted (Fritz, 1986). The overall effect is that charged chemical species do not move readily through a compact clay and neutral water molecules may be able to pass more freely.
It must be remembered that Donnan exclusion occurs in many systems other than “compact clay”. By instead considering e.g. a ferrocyanide solution, it becomes clear that salt exclusion has nothing to do with how hindered the ions are to move in the system (as long as they move). KCl is, of course, not excluded from a potassium ferrocyanide system because ferrocyanide repels chloride, nor does such interactions imply restricted mobility (repulsion occurs in all salt solutions). Similarly, salt is not excluded from bentonite because of repulsion between anions and surfaces (also, a negative potential does not repel anything — charge does).
In the above quotation it is easy to spot the flaw in the argument by switching roles of anions and cations; you may equally incorrectly say that cations are attracted, and that anions tag along in order to maintain charge neutrality.
The idea that “semi-permeability” (and “anion” exclusion) is caused by mobility restrictions for the ions within the bentonite, while water can “pass more freely” is found in many places in the bentonite literature. E.g. Shackelford and Moore (2013) write (where, again, potentials are described as repelling)
In [the case of bentonite], when the clay is compressed to a sufficiently high density such that the pore spaces between adjacent clay particles are minimized to the extent that the electrostatic (diffuse double) layers surrounding the particles overlap, the
overlapping negative potentials repel invading anions such that the pore becomes excluded to the anion. Cations also may be excluded to the extent that electrical neutrality in solution is required (e.g., Robinson and Stokes, 1959).
This phenomenon of anion exclusion also is responsible for the existence of semipermeable membrane behavior, which refers to the ability of a porous medium to restrict the migration of solutes, while allowing passage of the solvent (e.g., Shackelford, 2012).
Chagneau et al. (2015) write
[…] TOT layers bear a negative structural charge that is compensated by cation accumulation and anion depletion near their surfaces in a region known as the electrical double layer (EDL). This property gives clay materials their semipermeable
membrane properties: ion transport in the clay material is hindered by electrostatic repulsion of anions from the EDL porosity, while water is freely admitted to the membrane.
and Tournassat and Steefel (2019) write (where, again, we can switch roles of “co-” and “counter-ions”, to spot one of the flaws)
The presence of overlapping diffuse layers in charged nanoporous media is responsible for a partial or total repulsion of co-ions from the porosity. In the presence of a gradient of bulk electrolyte concentration, co-ion migration through the pores is hindered, as well as the migration of their counter-ion counterparts because of the electro-neutrality constraint. This explains the salt-exclusionary properties of these materials. These properties confer these media with a semi-permeable membrane behavior: neutral aqueous species and water are freely admitted through the membrane while ions are not, giving rise to coupled transport processes.
I am quite puzzled by these statements being so commonplace.3 It does not surprise me that all the quotations basically state some version of the incorrect notion that salt exclusion is caused by electrostatic repulsion between anions and surfaces — this is, for some reason, an established “explanation” within the clay literature.4 But all quotations also state (more or less explicitly) that ions (or even “solutes”) are restricted, while water can move freely in the clay. Given that one of the main features of compacted bentonite components is to restrict water transport, with hydraulic conductivities often below 10-13 m/s, I don’t really know what to say.
Furthermore, one of the most investigated areas in bentonite research is the (relatively) high cation transport capacity that can be achieved under the right conditions. In this light, I find it peculiar to claim that bentonite generally impedes ion transport in relation to water transport.
Bentonite as a non-ideal semi-permeable membrane
As far as I see, authors seem to confuse transport between external solutions and clay with processes that occur between two external solutions separated by a bentonite component. Here is an example of the latter set-up

The difference in concentration between the two solutions implies water transport — i.e. osmosis — from the reservoir with lower salt concentration to the reservoir with higher concentration. In this process, the bentonite component as a whole functions as the membrane.
The bentonite component has this function because in this process it is more permeable to water than to salt (which has a driving force to be transported from the high concentration to the low concentration reservoir). This is the sense in which bentonite can be said to be semi-permeable with respect to water/salt. Note:
- Salt is still transported through the bentonite. Thus, the bentonite component functions fundamentally only as a non-ideal membrane.
- Zooming in on the bentonite component in the above set-up, we note that the non-ideal semi-permeable functionality emerges from the presence of two ideal semi-permeable components. As discussed above, the ideal semi-permeable components (metal filters) keep the clay particles in place.

- The non-ideal semi-permeability is a consequence of salt exclusion. But these are certainly not the same thing! Rather, the implication is: Ideal semi-permeable components (impermeable to clay) \(\rightarrow\) Donnan effect \(\rightarrow\) Non-ideal semi-permeable membrane functionality (for salt)
- The non-ideal functionality means that it is only relevant during non-equilibrium. E.g., a possible (osmotic) pressure increase in the right compartment in the illustration above will only last until the salt has had time to even out in the two reservoirs; left to itself, the above system will eventually end up with identical conditions in the two reservoirs. This is in contrast to the effect of an ideal membrane, where it makes sense to speak of an equilibrium osmotic pressure.
- None of the above points depend critically on the membrane material being bentonite. The same principal functionality is achieved with any type of Donnan system. One could thus imagine replacing the bentonite and the metal filters with e.g. a ferrocyanide solution and appropriate ideal semi-permeable membranes. I don’t know if this particular system ever has been realized, but e.g. membranes based on polyamide rather than bentonite seems more commonplace in filtration applications (we have now opened the door to the gigantic fields of membrane and filtration technology). From this consideration it follows that “semi-permeability” cannot be attributed to anything bentonite specific (such as “overlapping double layers”, or direct interaction with charged surfaces).
- I think it is important to remember that, even if bentonite is semi-permeable in the sense discussed, the transfer of any substance across a compacted bentonite sample is significantly reduced (which is why we are interested in using it e.g. for confining waste). This is true for both water and solutes (perhaps with the exception of some cations under certain conditions).
“Semi-permeability” in experiments
Even if bentonite is not semi-permeable in the sense described in many places in the literature, its actual non-ideal semi-preamble functionality must often be considered in compacted clay research. Let’s have look at some relevant cases where a bentonite sample is separated by two external solution reservoirs.
Tracer through-diffusion
The simplest set-up of this kind is the traditional tracer through-diffusion experiment. Quite a lot of such tests have been published, and we have discussed various aspects of this research in earlier blog posts.

The traditional tracer through-diffusion test maintains identical conditions in the two reservoirs (the same chemical compositions and pressures) while adding a trace amount of the diffusing substance to the source reservoir. The induced tracer flux is monitored by measuring the amount of tracer entering the target reservoir.
In this case the chemical potential is identical in the two reservoirs for all components other than the tracer, and no additional transport processes are induced. Yet, it should be kept in mind that both the pressure and the electrostatic potential is different in the bentonite as compared with the reservoirs. The difference in electrostatic potential is the fundamental reason for the distinctly different diffusional behavior of cations and anions observed in these types of tests: as the background concentration is lowered, cation fluxes increase indefinitely (for constant external tracer concentration) while anion fluxes virtually vanish.
Tracer through-diffusion is often quantified using the parameter \(D_e\), defined as the ratio between steady-state flux and the external concentration gradient.5 \(D_e\) is thus a type of ion permeability coefficient, rather than a diffusion coefficient, which it nevertheless often is assumed to be.
Typically we have that \(D_e^\mathrm{cation} > D_e^\mathrm{water} > D_e^\mathrm{anion}\) (where \(D_e^\mathrm{cation}\) in principle may become arbitrary large). This behavior both demonstrates the underlying coupling to electrostatics, and that “charged chemical species” under these conditions hardly can be said to move less readily through the clay as compared with water molecules.
Measuring hydraulic conductivity
A second type of experiment where only a single component is transported across the clay is when the reservoirs contain pure water at different pressures. This is the typical set-up for measuring the so-called hydraulic conductivity of a clay component.6

Even if no other transport processes are induced (there is nothing else present to be transported), the situation is here more complex than for the traditional tracer through-diffusion test. The difference in water chemical potential between the two reservoirs implies a mechanical coupling to the clay, and a corresponding response in density distribution. An inhomogeneous density, in turn, implies the presence of an electric field. Water flow through bentonite is thus fundamentally coupled to both mechanical and electrical processes.
In analogy with \(D_e\), hydraulic conductivity is defined as the ratio between steady-state flow and the external pressure gradient. Consequently, hydraulic conductivity is an effective mass transfer coefficient that don’t directly relate to the fundamental processes in the clay.
An indication that water flow through bentonite is more subtle than what it may seem is the mere observation that the hydraulic conductivity of e.g. pure Na-montmorillonite at a porosity of 0.41 is only 8·10-15 m/s. This system thus contains more than 40% water volume-wise, but has a conductivity below that of unfractioned metamorphic and igneous rocks! At the same time, increasing the porosity by a factor 1.75 (to 0.72), the hydraulic conductivity increases by a factor of 75! (to 6·10-13 m/s7)
Mass transfer in a salt gradient
Let’s now consider the more general case with different chemical compositions in the two reservoirs, as well as a possible pressure difference (to begin with, we assume equal pressures).
Even with identical hydrostatic pressures in the reservoirs, this configuration will induce a pressure response, and consequently a density redistribution, in the bentonite. There will moreover be both an osmotic water flow from the right to the left reservoir, as well as a diffusive solute flux in the opposite direction. This general configuration thus necessarily couples hydraulic, mechanical, electrical, and chemical processes.
This type of configuration is considered e.g. in the study of osmotic effects in geological settings, where a clay or shale formation may act as a membrane.8 But although this configuration is highly relevant for engineered clay barrier systems, I cannot think of very many studies focused on these couplings (perhaps I should look better).
For example, most through-diffusion studies are of the tracer type discussed above, although evaluated parameters are often used in models with more general configurations (e.g. with salt or pressure gradients). Also, I am not aware of any measurements of hydraulic conductivity in case of a salt gradient (but the same hydrostatic pressure), and I am even less aware of such values being compared with those evaluated in conventional tests (discussed previously).
A quite spectacular demonstration that mass transfer may occur very differently in this general configuration is the seeming steady-state uphill diffusion effect: adding an equal concentration of a cation tracer to the reservoirs in a set-up with a maintained difference in background concentration, a tracer concentration difference spontaneously develops. \(D_e\) for the tracer can thus equal infinity,9 or be negative (definitely proving that this parameter is not a diffusion coefficient). I leave it as an exercise to the reader to work out how “semi-permeable” the clay is in this case. Update (240822): The “uphill” diffusion effect is further discussed here.
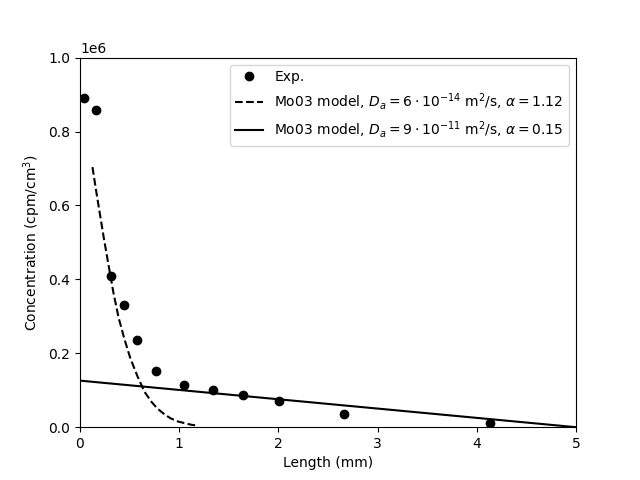
A process of practical importance for engineered clay barrier systems is hyperfiltration of salts. This process will occur when a sufficient pressure difference is applied over a bentonite sample contacted with saline solutions. Water and salt will then be transferred in the same direction, but, due to exclusion, salt will accumulate on the inlet side. A steady-state concentration profile for such a process may look like this

The local salt concentration at the sample interface on the inlet side may thus be larger than the concentration of the injected solution. This may have consequences e.g. when evaluating hydraulic conductivity using saline solutions.
Hyperfiltration may also influence the way a sample becomes saturated, if saturated with a saline solution. If the region near the inlet is virtually saturated, while regions farther into the sample still are unsaturated, hyperfiltration could occur. In such a scenario the clay could in a sense be said to be semi-permeable (letting through water and filtrating salts), but note that the net effect is to transfer more salt into the sample than what is dictated by Donnan equilibrium with the injected solution (which has concentration \(c_1\), if we stick with the figure above). Salt will then have to diffuse out again, in later stages of the process, before full equilibrium is reached. This is in similarity with the saturation process that we considered earlier.
Footnotes
[1] We have considered this study before, when discussing the empirical evidence for salt in interlayers.
[2] This is more than a thought-experiment; a test just like this was conducted by Karnland et al. (2005). Here is the recorded pressure response of a Na-montmorillonite sample (dry density 1.4 g/cm3) as it is contacted with NaCl solutions of increasing concentration

We have considered this study earlier, as it proves that salt enters interlayers.
[3] As a side note, is the region near the surface supposed to be called “diffuse layer”, “electrical double layer”, or “electrostatic (diffuse double) layer”?
[4] Also Fritz (1986), referenced in the quotation by Horseman et al. (1996), states a version of this “explanation”.
[5] This is not a gradient in the mathematical sense, but is defined as \( \left (c_\mathrm{target} – c_\mathrm{source} \right)/L\), where \(L\) is sample length.
[6] Hydraulic conductivity is often also measured using a saline solution, which is commented on below.
[7] Which still is an a amazingly small hydraulic conductivity, considering the the water content.
[8] The study of Neuzil (2000) also provides clear examples of water moving out of the clay, and salt moving in, in similarity with the process considered above.
[9] Mathematically, the statement “equal infinity” is mostly nonsense, but I am trying to convey that a there is a tracer flux even without any external tracer concentration difference.









{kind=link}