
On the surface, “Ionendiffusion in Hochverdichtetem Bentonit”1 by G. Kahr, R. Hasenpatt, and M. Müller-Vonmoos, published by NAGRA in March 1985, looks like an ordinary mundane 37-page technical report. But it contains experimental results that could have completely changed the history of model development for compacted clay.
Test principles
The tests were conducted in a quite original manner. By compacting granules or powder, the investigators obtained samples that schematically look like this

The bentonite material — which was either Na-dominated “MX-80”, or Ca-dominated “Montigel” — was conditioned to a specific water-to-solid mass ratio \(w\). At one of the faces, the bentonite was mixed with a salt (in solid form) to form a thin source for diffusing ions. This is essentially the full test set-up! Diffusion begins as soon as the samples are prepared, and a test was terminated after some prescribed amount of time, depending on diffusing ion and water content. At termination, the samples were sectioned and analyzed. In this way, the investigators obtained final state ion distributions, which in turn were related to the initial states by a model, giving the diffusion coefficients of interest.
Note that the experiments were conducted without exposing samples to a liquid (external) solution; the samples were “unsaturated” to various degree, and the diffusing ions dissolve within the bentonite. The samples were not even confined in a test cell, but “free-standing”, and consequently not under pressure. They were, however, stored in closed vessels during the course of the tests, to avoid changes in water content.
With this test principle a huge set of diffusion tests were performed, with systematic variation of the following variables:
- Bentonite material (“MX-80” or “Montigel”)
- Water-to-solid mass ratio (7% — 33%)
- Dry density (1.3 g/m3 — 2.1 g/m3 )
- Diffusing salt (SrCl2, SrI2, CsCl, CsI, UO2(NO3)2, Th(NO3)4, KCl, KI, KNO3, K2SO4, K2CO3, KF)
Distribution of water in the samples
From e.g. X-ray diffraction (XRD) we know that bentonite water at low water content is distributed in distinct, sub-nm thin films. For simplicity we will refer to all water in the samples as interlayer water, although some of it, reasonably, forms interfaces with air. The relevant point is that the samples contain no bulk water phase, but only interfacial (interlayer) water.
I argue extensively on this blog for that interlayer water is the only relevant water phase also in saturated samples under pressure. In the present case, however, it is easier to prove that this is the case, as the samples are merely pressed bentonite powder at a certain water content; the bentonite water is not pressurized, the samples are not exposed to liquid bulk water, nor are they in equilibrium with liquid bulk water. Since the water in the samples obviously is mobile — as vapor, but most reasonably also in interconnected interlayers — it is a thermodynamic consequence that it distributes as to minimize the chemical potential.
There is a ton of literature on how the montmorillonite basal spacing varies with water content. Here, we use the neat result from Holmboe et al. (2012) that the average interlayer distance varies basically linearly2 with water content, like this

XRD-studies also show that bentonite water is distributed in rather distinct hydration states, corresponding to 0, 1, 2, or 3 monolayers of water.3 We label these states 0WL, 1WL, 2WL, and 3WL, respectively. In the figure is indicated the approximate basal distances for pure 1WL (12.4 Å), 2WL (15.7 Å), and 3WL (19.0 Å), which correspond roughly to water-to-solid mass ratios of 0.1, 0.2, and 0.3, respectively.
From the above plot, we estimate roughly that the driest samples in Kahr et al. (1985) (\(w \sim 0.1\)) are in pure 1WL states, then transitions to a mixture of 1WL and 2WL states (\(w\sim 0.1 – 0.2\)), to pure 2WL states (\(w \sim 0.2\)), to a mixture of 2WL and 3WL states (\(w\sim 0.2 – 0.3\)), and finally to pure 3WL states (\(w\sim 0.3\)).
Results
With the knowledge of how water is distributed in the samples, let’s take a look at the results of Kahr et al. (1985).
Mobility of interlayer cations confirmed
The most remarkable results are of qualitative character. It is, for instance, demonstrated that several cations diffuse far into the samples. Since the samples only contain interlayer water, this is a direct proof of ion mobility in the interlayers!
Also, cations are demonstrated to be mobile even when the water content is as low as 7 or 10 %! As such samples are dominated by 1WL states, this is consequently evidence for ion mobility in 1WL states.
A more quantitative assessment furthermore shows that the cation diffusivities varies with water content in an almost step-wise manner, corresponding neatly to the transitions between various hydration states. Here is the data for potassium and strontium

This behavior further confirms that the ions diffuse in interlayers, with an increasing diffusivity as the interlayers widen.
It should also be noted that the evaluated values of the diffusivities are comparable to — or even larger4 — than corresponding results from saturated, pressurized tests. This strongly suggests that interlayer diffusivity dominates also in the latter types of tests, which also has been confirmed in more recent years. The larger implication is that interlayer diffusion is the only relevant type of diffusion in general in compacted bentonite.
Anions enter interlayers (and are mobile)
The results also clearly demonstrate that anions (iodide) diffuse in systems with water-to-solid mass ratio as low as 7%! With no other water around, this demonstrates that anions diffuse in — and consequently have access to — interlayers. This finding is strongly confirmed by comparing the \(w\)-dependence of diffusivity for anions and cations. Here is plotted the data for iodide and potassium (with the potassium diffusivity indicated on the right y-axis)
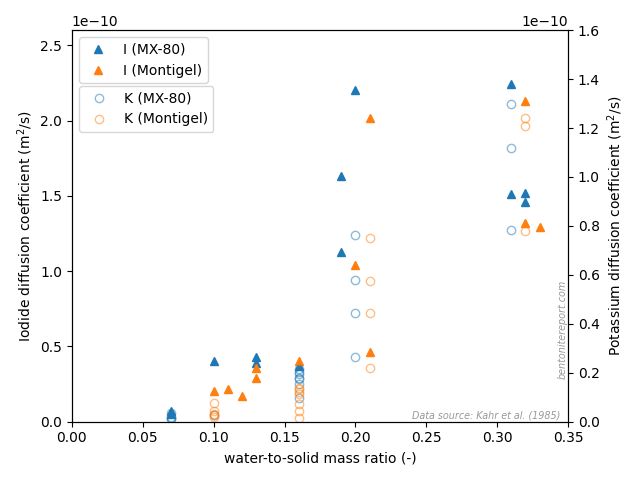
The iodide mobility increases as the system transitions from 1WL to 2WL, in a very similar way as for potassium (and strontium). If this is not a proof that the anion diffuse in the same domain as the cation I don’t know what is! Also for iodide the value of the diffusivity is comparable to what is evaluated in water saturated systems under pressure, which implies that interlayer diffusivity dominates generally in compacted bentonite, also for anions.
Dependence of diffusivity on water content and density
A conclusion made in Kahr et al. (1985), that I am not sure I fully agree with, is that diffusivity mainly depends on water content rather than density. As seen in the diagrams above, the spread in diffusivity is quite substantial for a given value of \(w\). There is actually some systematic variation here: for constant \(w\), diffusivity tend to increase with dry density.
Although using unsaturated samples introduces additional variation, the present study provides a convenient procedure to study diffusion in systems with very low water content. A more conventional set-up in this density limit has to deal with enormous pressures (on the order of 100 MPa).
Interlayer chemistry
An additional result is not acknowledged in the report, but is a direct consequence of the observations: the tests demonstrate that interlayers are chemically active. The initially solid salt evidently dissolves before being able to diffuse. Since these samples are not even close to containing a bulk water phase (as discussed above), the dissolution process must occur in an interlayer. More precisely, the salt must dissolve in interface water between the salt mineral and individual montmorillonite layers, as illustrated here

This study seems to have made no impact at all
In the beginning of 1985, the research community devoted to radioactive waste barriers seems to have been on its way to correctly identify diffusion in interlayers as the main transport mechanism, and to recognize how ion diffusion in bentonite is influenced by equilibrium with external solutions.
Already in 1981, Torstenfelt et al. (1981) concluded that the traditional diffusion-sorption model is not valid, for e.g. diffusion of Sr and Cs, in compacted bentonite. They also noted, seemingly without realizing the full importance, that these ions diffused even in unsaturated samples with as low water-to-solid mass ratio as 10%.
A significant diffusion was observed for Sr in dry clay, although slower than for water saturated clay, Figure 4, while Cs was almost immobile in the dry clay.
A year later also Eriksen and Jacobsson (1982) concluded that the traditional diffusion model is not valid. They furthermore pointed out the subtleties involved when interpreting through-diffusion experiments, due to ion equilibrium effects
One difficulty in correlating the diffusivities obtained from profile analysis to the diffusivities calculated from steady state transport data is the lack of knowledge of the tracer concentration at the solution-bentonite interface. This concentration is generally higher for sorbing species like positive ions (counterions to the bentonite) and lower for negative ions (coions to the bentonite) as shown schematically in figure 11. The equilibrium concentration of any ion in the bentonite and solution respectively is a function of the ionic charge, the ionic strength of the solution and the overall exchanger composition and thereby not readily calculated
In Eriksen and Jacobsson (1984) the picture is fully clear
By regarding the clay-gel as a concentrated electrolytic system Marinsky has calculated (30) distribution coefficients for Sr2+ and Cs+ ions in good agreement with experimentally determined Kd-values. The low anionic exchange capacity and hence the low anion concentration in the pore solution caused by Donnan exclusion also explain the low concentrations of anionic tracers within the clay-gel
[…]
For simple cations the ion-exchange process is dominating and there is, as also pointed out by Marinsky (30), no need to suppose that the counterions are immobilized. It ought to be emphasized that for the compacted bentonite used in the diffusion experiments discussed in this report the water content corresponds roughly to 2-4 water molecule layers (31). There is therefore really no “free water” and the measured diffusivity \(\bar{D}\) can be regarded as corresponding approximately to the diffusivity within the adsorbed phase […]
Furthermore, also Soudek et al. (1984) had discarded the traditional diffusion-sorption model, identified the exchangeable cations as giving a dominating contribution to mass transfer, and used Donnan equilibrium calculations to account for the suppressed internal chloride concentration.
In light of this state of the research front, the contribution of Kahr et al. (1985) cannot be described as anything but optimal. In contrast to basically all earlier studies, this work provides systematic variation of several variables (most notably, the water-to-solid ratio). As a consequence, the results provide a profound confirmation of the view described by Eriksen and Jacobsson (1984) above, i.e. that interlayer pores essentially govern all physico-chemical behavior in compacted bentonite. A similar description was later given by Bucher and Müller-Vonmoos (1989) (though I don’t agree with all the detailed statements here)
There is no free pore water in highly compacted bentonite. The water in the interlayer space of montmorillonite has properties that are quite different from those of free pore water; this explains the extremely high swelling pressures that are generated. The water molecules in the interlayer space are less mobile than their free counterparts, and their dielectric constant is lower. The water and the exchangeable cations in the interlayer space can be compared to a concentrated salt solution. The sodium content of the interlayer water, at a water content of 25%, corresponds approximately to a 3-n salt solution, or six times the concentration in natural seawater. This more or less ordered water is fundamentally different from that which engineers usually take into account; in the latter case, pore water in a saturated soil is considered as a freely flowing fluid. References to the porosity in highly compacted bentonite are therefore misleading. Highly compacted bentonite is an unfamiliar material to the engineer.
Given this state of the research field in the mid-80s, I find it remarkable that history took a different turn. It appears as the results of Kahr et al. (1985) made no impact at all (it may be noticed that they themselves analyzed the results in terms of the traditional diffusion-sorption model). And rather than that researchers began identifying that transport in interlayers is the only relevant contribution, the so-called surface diffusion model gained popularity (it was already promoted by e.g. Soudek et al. (1984) and Neretnieks and Rasmuson (1983)). Although this model emphasizes mobility of the exchangeable cations, it is still centered around the idea that compacted bentonite contains bulk water.5 Most modern bentonite models suffer from similar flaws: they are formulated in terms of bulk water, while many effects related to interlayers are treated as irrelevant or optional.
For the case of anion diffusion the historical evolution is maybe even more disheartening. In 1985 the notions of “effective” or “anion-accessible” porosities seem to not have been that widely spread, and here was clear-cut evidence of anions occupying interlayer pores. But just a few years later the idea began to grow that the pore space in compacted bentonite should be divided into regions which are either accessible or inaccessible to anions. As far as I am aware, the first use of the term “effective porosity” in this context was used by Muurinen et al. (1988), who, ironically, seem to have misinterpreted the Donnan equilibrium approach presented by Soudek et al. (1984). To this day, this flawed concept is central in many descriptions of compacted clay.
Footnotes
[1] “Ion diffusion in highly compacted bentonite”
[2] Incidentally, the slope of this line corresponds to a water “density” of 1.0 g/cm3.
[3] This is the region of swelling often referred to as “crystalline”.
[4] I’m not sure the evaluation in Kahr et al. (1985) is fully correct. They use the solution to the diffusion equation for an impulse source (a Gaussian), but, to my mind, the source is rather one of constant concentration (set by the solubility of the salt). Unless I have misunderstood, the mathematical expression to be fitted to data should then be an erfc-function, rather than a Gaussian. Although this modification would change the numerical values of the evaluated diffusion coefficients somewhat, it does not at all influence the qualitative insights provided by the study.
[5] I have discussed the surface diffusion model in some detail in previous blog posts.