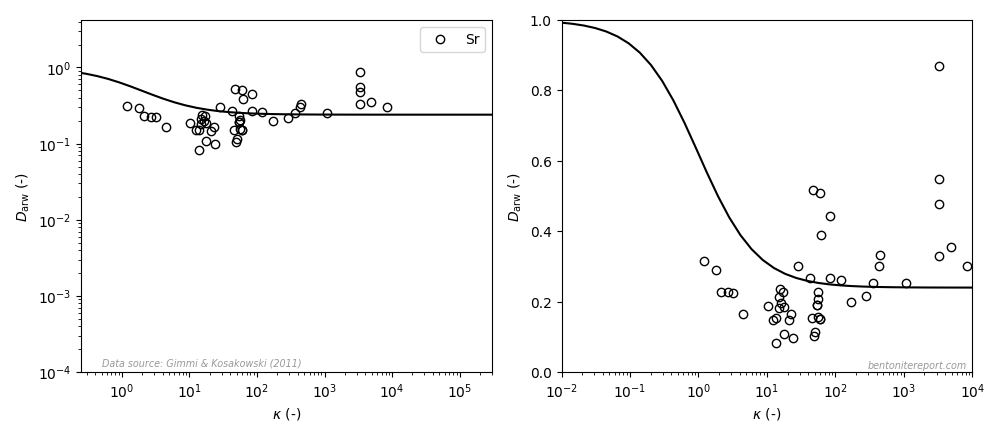
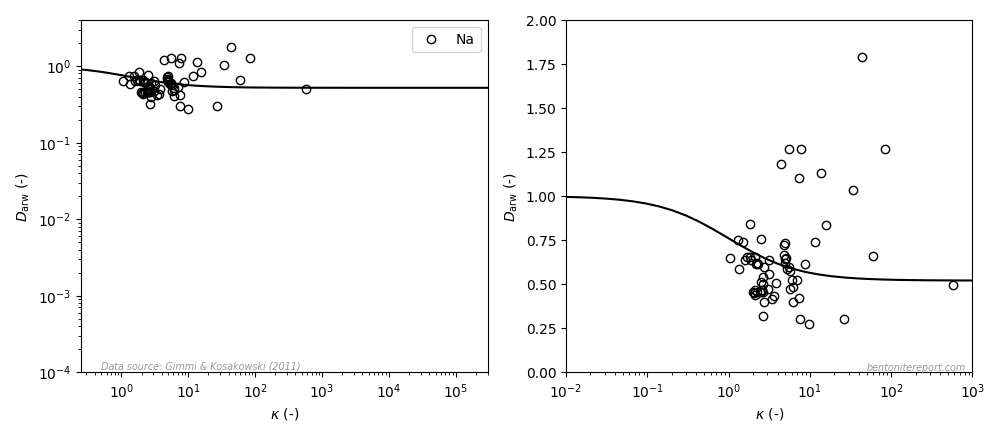
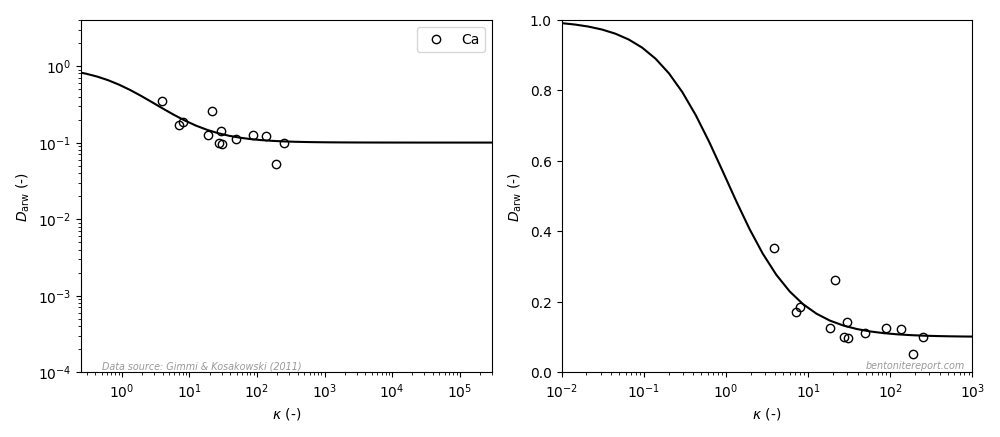
Recently, we discussed reported equilibrium chloride concentrations in sodium dominated bentonite, and identified a need to assess the individual studies. As most data is obtained from through-diffusion experiments, we here take a general look at how anion equilibrium is a part of the through-diffusion set-up, and how we can use reported model parameters to extract the experimentally accessible equilibrium concentrations.
We define the experimentally accessible concentration of a chemical species in a bentonite sample as
\begin{equation} \bar{c} = \frac{n}{m_\mathrm{w}} \end{equation}
where \(n\) is the total amount of the species,1 and \(m_{w}\) is the total water mass in the clay.2 It should be clear that \(\bar{c}\), which we will refer to as the clay concentration, is accessible without relying on any particular model concept.
An equilibrium concentration is defined as the corresponding clay concentration (i.e. \(\bar{c}\)) of a species when the clay is in equilibrium with an external solution with species concentration \(c^\mathrm{ext}\). A convenient way to express this equilibrium is in terms of the ratio \(\bar{c}/c^\mathrm{ext}\).
The through-diffusion set-up
A through-diffusion set-up consists of a (bentonite) sample sandwiched between a source and a target reservoir, as illustrated schematically here (for some arbitrary time):

The sample length is labeled \(L\), and we assume the sample to be initially empty of the diffusing species. A test is started by adding a suitable amount of the diffusing species to the source reservoir. Diffusion through the bentonite is thereafter monitored by recording the concentration evolution in the target reservoir,3 giving an estimation of the flux out of the sample (\(j^\mathrm{out}\)). The clay concentration for anions is typically lower than the corresponding concentration in the source reservoir.
Although a through-diffusion test is not in full equilibrium (by definition), local equilibrium prevails between clay and external solution4 at the interface to the source reservoir (\(x=0\)). Thus, even if the source concentration varies, we expect the ratio \(\bar{c}(0)/c^\mathrm{source}\) to stay constant during the course of the test.5
The effective porosity diffusion model
Our primary goal is to extract the concentration ratio \(\bar{c}(0)/c^\mathrm{source}\) from reported through-diffusion parameters. These parameters are in many anion studies specific to the “effective porosity” model, rather than being accessible directly from the experiments. We therefore need to examine this particular model.
The effective porosity model divides the pore space into a bulk water domain and a domain that is assumed inaccessible to anions. The porosity of the bulk water domain is often referred to as the “effective” or the “anion-accessible” porosity, and here we label it \(\epsilon_\mathrm{eff}\).
Anions are assumed to diffuse in the bulk water domain according to Fick’s first law
\begin{equation} \label{eq:Fick1_eff} j = -\epsilon_\mathrm{eff} \cdot D_p \cdot \nabla c^\mathrm{bulk} \tag{1} \end{equation}
where \(D_p\) is the pore diffusivity in the bulk water phase. This relation is alternatively expressed as \(j = -D_e \cdot \nabla c^\mathrm{bulk}\), which defines the effective diffusivity \(D_e = \epsilon_\mathrm{eff} \cdot D_p\).
Diffusion is assumed to be the only mechanism altering the concentration, leading to Fick’s second law
\begin{equation} \label{eq:Fick2_eff} \frac{\partial c^\mathrm{bulk}}{\partial t} = D_p\cdot \nabla^2 c^\mathrm{bulk} \tag{2} \end{equation}
Connection with experimentally accessible quantities
The bulk water concentration in the effective porosity model relates to the experimentally accessible concentration as
\begin{equation} \label{eq:cbar_epsilon} \bar{c} = \frac{\epsilon_\mathrm{eff}}{\phi} c^\mathrm{bulk} \tag{3} \end{equation}
where \(\phi\) is the physical porosity of the sample. Since a bulk water concentration varies continuously across interfaces to external solutions, we have \(c^\mathrm{bulk}(0) = c^\mathrm{source}\) at the source reservoir, giving
\begin{equation} \label{eq:cbar_epsilon0} \frac{\bar{c}(0)}{c^\mathrm{source}} = \frac{\epsilon_\mathrm{eff}} {\phi} \tag{4} \end{equation}
This equation shows that the effective porosity parameter quantifies the anion equilibrium concentration that we want to extract. That is not to say that the model is valid (more on that later), but that we can use eq. 4 to translate reported model parameters to an experimentally accessible quantity.
In principle, we could finish the analysis here, and use eq. eq. 4 as our main result. But most researchers do not evaluate the effective porosity in the direct way suggested by this equation (they may not even measure \(\bar{c}\)). Instead, they evaluate \(\epsilon_\mathrm{eff}\) from a fitting procedure that also includes the diffusivity as a parameter. It is therefore fruitful to also include the transport aspects of the through-diffusion test in our analysis.
From closed-cell diffusion tests, we know that the clay concentration evolves according to Fick’s second law, both for many cations and anions. We will therefore take as an experimental fact that \(\bar{c}\) evolves according to
\begin{equation} \label{eq:Fick2_exp} \frac{\partial \bar{c}}{\partial t} = D_\mathrm{macr.} \nabla^2 \bar{c} \tag{5} \end{equation}
This equation defines the diffusion coefficient \(D_\mathrm{macr.}\), which should be understood as an empirical quantity.
Combining eqs. 3 and 2 shows that \(D_p\) governs the evolution of \(\bar{c}\) in the effective porosity model (if \(\epsilon_\mathrm{eff}/\phi\) can be considered a constant). A successful fit of the effective porosity model to experimental data thus provides an estimate of \(D_\mathrm{macr.}\) (cf. eq. 5), and we may write
\begin{equation} D_p = D_\mathrm{macr.} \tag{6} \end{equation}
With the additional assumption of constant reservoir concentrations, eq. 2 has a relatively simple analytical solution, and the corresponding outflux reads
\begin{equation} \label{eq:flux_analytic} j^\mathrm{out}(t) = j^\mathrm{ss} \left ( 1 + 2\sum_{n=1}^\infty \left (-1 \right)^n e^{-\frac{\pi^2n^2 D_\mathrm{p} t}{L^2}} \right ) \tag{7} \end{equation}
where \(j^\mathrm{ss}\) is the steady-state flux. In steady-state, \(c^\mathrm{bulk}\) is distributed linearly across the sample, and we can express the gradient in eq. 1 using the reservoir concentrations, giving
\begin{equation} j^\mathrm{ss} = \epsilon_\mathrm{eff} \cdot D_\mathrm{p} \cdot \frac{c^\mathrm{source}}{L} \tag{8} \end{equation}
where we have assumed zero target concentration.
Treating \(j^\mathrm{ss}\) as an empirical parameter (it is certainly accessible experimentally), and using eq. 6, we get another expression for \(\epsilon_\mathrm{eff}\) in terms of experimentally accessible quantities
\begin{equation} \epsilon_\mathrm{eff} = \frac{j^\mathrm{ss}\cdot L}{c^\mathrm{source} \cdot D_\mathrm{macr.} } \tag{9} \end{equation}
This relation (together with eqs. 4 and 6) demonstrates that if we fit eq. 7 using \(D_p\) and \(j^\mathrm{ss}\) as fitting parameters, the equilibrium relation we seek is given by
\begin{equation} \label{eq:exp_estimate} \frac{\bar{c}(0)}{c^\mathrm{source}} = \frac{j^\mathrm{ss}\cdot L} {\phi \cdot c^\mathrm{source} \cdot D_\mathrm{macr.} } \tag{10} \end{equation}
This procedure may look almost magical, since any explicit reference to the effective porosity model has now disappeared; eq. 10 can be viewed as a relation involving only experimentally accessible quantities.
But the validity of eq. 10 reflects the empirical fact that the (steady-state) flux can be expressed using the gradient in \(\bar{c}\) and the physical porosity. The effective porosity model can be successfully fitted to anion through-diffusion data simply because it complies with this fact. Consequently, a successful fit does not validate the effective porosity concept, and essentially any description for which the flux can be expressed as \(j = -\phi\cdot D_p \cdot \nabla\bar{c}\) will be able to fit to the data.
We may thus consider a generic model for which eq. 5 is valid and for which a steady-state flux is related to the external concentration difference as
\begin{equation} \label{eq:jss_general} j_\mathrm{ss} = – \beta\cdot D_p \cdot \frac{c^\mathrm{target} – c^\mathrm{source}}{L} \tag{11} \end{equation}
where \(\beta\) is an arbitrary constant. Fitting such a model, using \(\beta\) and \(D_p\) as parameters, will give an estimate of \(\bar{c}(0)/c^\mathrm{source}\) (\(=\beta / \phi\)).
Note that the system does not have to reach steady-state — eq. 11 only states how the model relates a steady-state flux to the reservoir concentrations. Moreover, the model being fitted is generally numerical (analytical solutions are rare), and may account for e.g. possible variation of concentrations in the reservoirs, or transport in the filters connecting the clay and the external solutions.
The effective porosity model emerges from this general description by interpreting \(\beta\) as quantifying the volume of a bulk water phase within the bentonite sample. But \(\beta\) can just as well be interpreted e.g. as an ion equilibrium coefficient (\(\phi\cdot \Xi = \beta\)), showing that this description also complies with the homogeneous mixture model.
Additional comments on the effective porosity model
The effective porosity model can usually be successfully fitted to anion through-diffusion data (that’s why it exists). The reason is not because the data behaves in a manner that is difficult to capture without assuming that anions are exclusively located in a bulk water domain, but simply because this model complies with eqs. 5 and 11. We have seen that also the homogeneous mixture model — which makes the very different choice of having no bulk water at all within the bentonite — will fit the data equally well: the two fitting exercises are equivalent, connected via the parameter identification \(\epsilon_\mathrm{eff} \leftrightarrow \phi\cdot\Xi\).
Given the weak validation of the effective porosity model, I find it concerning that most anion through-diffusion studies are nevertheless reported in a way that not only assumes the anion-accessible porosity concept to be valid, but that treats \(\epsilon_\mathrm{eff}\) basically as an experimentally measured quantity.
Perhaps even more remarkable is that authors frequently treat the effective porosity model as were it some version of the traditional diffusion-sorption model. This is often done by introducing a so-called rock capacity factor \(\alpha\) — which can take on the values \(\alpha = \phi + \rho\cdot K_d\) for cations, and \(\alpha = \epsilon_\mathrm{eff}\) for anions — and write \(D_e = \alpha D_a\), where \(D_a\) is the “apparent” diffusion coefficient. The reasoning seems to go something like this: since the parameter in the governing equation in one model can be written as \(D_e/\epsilon_\mathrm{eff}\), and as \(D_e/(\phi + \rho\cdot K_d)\) in the other, one can view \(\epsilon_\mathrm{eff}\) as being due to negative sorption (\(K_d < 0\)).
But such a mixing of completely different mechanisms (volume restriction vs. sorption) is just a parameter hack that throws most process understanding out the window! In particular, it hides the fact that the effective porosity and diffusion-sorption models are incompatible: their respective bulk water domains have different volumes. Furthermore, this lumping together of models has led to that anion diffusion coefficients routinely are reported as “apparent”, although they are not; the underlying model contains a pore diffusivity (eq. 2). As I have stated before, the term “apparent” is supposed to convey the meaning that what appears as pure diffusion is actually the combined result of diffusion, sorption, and immobilization. Sadly, in the bentonite literature, “apparent diffusivity” often means “actual diffusivity”.
Footnotes
[1] For anions, the total amount is relatively easy to measure by e.g. aqueous extraction. Cations, on the other hand, will stick to the clay, and need to be exchanged with some other type of cation (not initially present). In any case, the total amount of a species (\(n\)) can in principle be obtained experimentally, in an unambiguous manner.
[2] Another reasonable choice would be to divide by the total sample volume.
[3] If the test is designed as to have a significant change of the source concentration, it is a good idea to also measure the concentration evolution in this reservoir.
[4] Here we assume that the transfer resistance of the filter is negligible.
[5] Provided that the rest of the aqueous chemistry remains constant, which is not always the case. For instance, cation exchange may occur during the course of the test, if the set-up involves more than one type of cation, and there may be ongoing mineral dissolution.